








转自:生信菜鸟团(微信公众号)
文献速报
主题 :Molecular Cancer 杂志 (7.776) 最新简报
文章于19年1月10日发表在 Molecular Cancer 上,文章简单分析了三位结肠直肠癌 (CRC) 患者以及三位 结直肠癌肝转移 (CRC-m) 患者样本中circRNA表达情况。

去核糖体、富集circRNA文库,HiSeq 4000 PE获得数据,STAR用于比对,DCC 用于circRNA的预测,使用 circBase 及 circ2Trait 疾病数据库对circRNA进行注释。差异circRNA通过定量PCR实验进行验证,并预测靶miRNA。然后就没有了…
所以说简单分析,文章可能的亮点在于,使用了转移的癌症样本,并引入 Student’s t-test及 Mann-Whitney test对独立样本间circRNA进行分析,并作 ROC曲线评估 circRNA 潜在的诊断价值。分析方法有一定参考价值。
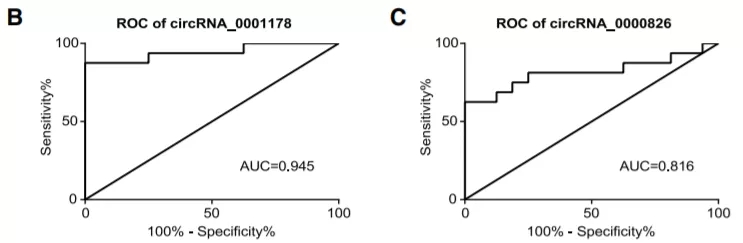
第二篇文章同样发表于 Molecular Cancer,发表于19年1月8日。作者挑取四对三阴性乳腺癌(TNBC)组织及癌旁组织,去核糖体后建库 HiSeq2500 平台测序,差异circRNA及mRNA表达分析获得一条上调的 circAGFG1 (15.04 folds) 作为验证对象。通过过表达、荧光原位杂交、双荧光素酶报告基因、RNA免疫沉淀、pull-down、western-blot、细胞周期、侵染及动物转染等实验验证了circAGFG1 与 miR-195-5p 之间的互作关系,并作调控通路图。

相较于第一篇来说,工作量是碾压级别的,除了过表达实验外,还对应在小鼠上进行验证。同时将验证的circRNA与临床病理特征进行相关系分析以及单因素、多因素回归分析。论证 circAGFG1 是一种潜在的预后标志物,具有诊断及治疗靶点的应用价值。
上一期我们介绍了一个上线不久的数据库 circBank,circBank提供一套更易读的 circRNA 名称,并支持与 circBase ID互换。 circBank 包含更详细人类circRNA,如编码位点预测、IRES元件预测及评估、变异位点以及m6A信息,且提供靶miRNA预测软件选择,其搜索框支持circBase ID,忽略大小写,但是不支持模糊搜索,需要注意。
今天的推送我们将推送一款鉴定circRNA的程序,由circRNA权威课题组中科院陈玲玲及杨力研究组开发维护,目前已更新到 CIRCexplorer2,除了预测circRNA外,能够探测可变剪切事件,且支持De Novo Assembly。还提供一些好用的 python 脚本,值得一看。
前期准备
CIRCexplorer2安装可以使用一下方法,安装过程是会将依赖的python包一并安装。
# PyPI
$ pip install circexplorer2
# Bioconda
$ conda install circexplorer2
# github
$ git clone https://github.com/YangLab/CIRCexplorer2.git
$ cd CIRCexplorer2
$ pip install -r requirements.txt
# install scipy according to http://www.scipy.org/install.html
$ python setup.py install
同时作者在这里提供一个抓取 UCSC数据库上 基因组、注释文件的小脚本,使用方法如下:
$ fetch_ucsc.py hg19/hg38/mm9/mm10 ref/kg/ens/fa <out文件>
下载的 RefSeq, KnownGenes or Ensembl注释文件需要转换为gtf文件,包含如下信息:

转化文件依赖 genePredToGtf,举一个栗子:
$ fetch_ucsc.py hg19 ref hg19_ref.txt
$ cut -f2-11 hg19_ref.txt|genePredToGtf file stdin hg19_ref.gtf
分析流程
虽然在 CIRCexplorer2的帮助文档中提到:
Although different aligners showed slight difference in circular RNA identification, TopHat2/TopHat-Fusion has a perfect match with Cufflinks.
目前,大多数该软件的推文都基于TopHat2/TopHat-Fusion流程,但是TopHat2/TopHat-Fusion 几乎已被完全替代。因此,我们今天的流程将从支持的比对软件 STAR 展开。 STAR是一款 ENCODE计划的御用软件,在17年 Nature Communications 发表RNA-seq分析软件比较中, STAR 较 TopHat 和 HASAT2来说,具有较高的唯一比对率 (highest fraction of uniquely mapped read pairs),对错配具有较高的容忍度。
# 安装
$ wget https://github.com/alexdobin/STAR/archive/2.5.3a.tar.gz
$ tar -xzf 2.5.3a.tar.gz && cd STAR-2.5.3a
# 建索引
$ STAR --runThreadN 8 --runMode genomeGenerate --genomeDir <index输出路径> --genomeFastaFiles <基因组序列> --sjdbGTFfile <参考基因组注释文件>
# PE比对
$ STAR --chimSegmentMin 10 --runThreadN 8 --genomeDir <index文件> --readFilesIn <read_1.fastq read_2.fastq> --outFileNamePrefix <输出文件(默认bam文件)>
# parsing,得到back_spliced_junction.bed
$ CIRCexplorer2 parse -t STAR Chimeric.out.junction > CIRCexplorer2_parse.log
# Annotating,得到circularRNA_known.txt
$ CIRCexplorer2 annotate -r <基因注释文件> -g <基因组文件> -b back_spliced_junction.bed -o circularRNA_known.txt > CIRCexplorer2_annotate.log
最终的文件内容将包含以下信息:
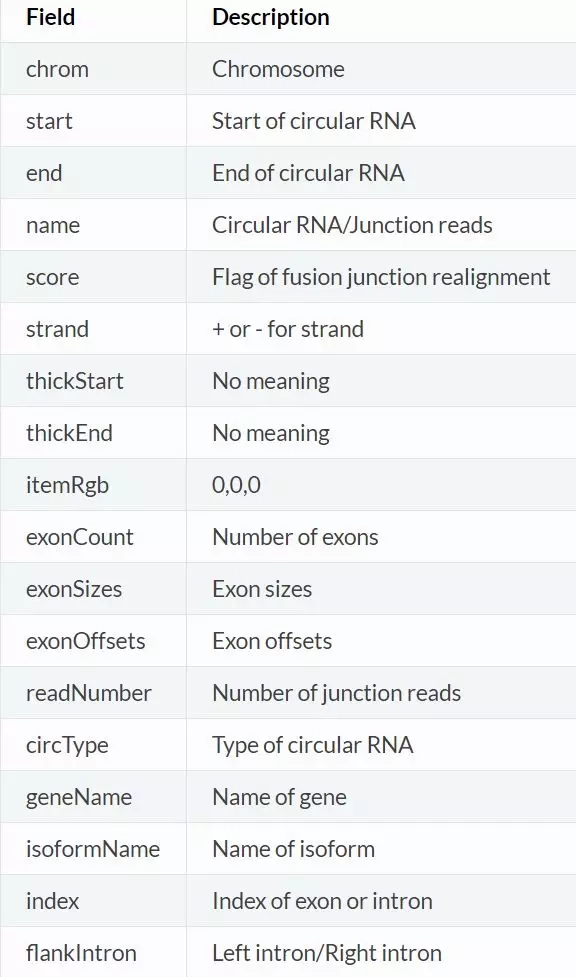
我们能够查看 circularRNA_known.txt 获得鉴定结果,除了一些基础数据的统计外,我们可以根据 circ_fusion.txt文件中提供的 circRNA 位置使用 bedtools getfasta提取序列文件后,结合差异表达,进行靶miRNA预测等分析。
小番外
CIRCexplorer2支持 De Novo Assembly,基于tophat比对Cufflinks 组装,支持poly(A)−/ribo− RNA-seq。最终将产生三个文件夹。代码如下:
$ CIRCexplorer2 assemble -r hg19_ref_all.txt -m tophat -o assemble > CIRCexplorer2_assemble.log
$ CIRCexplorer2 denovo -r hg19_ref_all.txt -g hg19.fa -b back_spliced_junction.bed --abs abs --as as -m tophat -n pAplus_tophat -o denovo > CIRCexplorer2_denovo.log
支持De Novo Assembly获得全长 circRNA 转录本的 circRNA 预测程序并不多,同时包含 circRNA 形成可变剪切的预测,若有稀有物种的探索,可以一试。
来第一个抢占沙发评论吧!