








转自:生信菜鸟团(微信公众号)
文献速报

作者基于人类的血管平滑肌细胞 (VSMC) circRNA的研究,通过小鼠模型,并通过进一步验证了circ_Lrp6 - miRNA-145z之间的互作,并对相关疾病进行调查,指出在不同的条件下circ_Lrp6和miRNA-145在小鼠和人类疾病的血管病变中受到不同的调节,并且circ_Lrp6的调节可能影响血管疾病的发展。文章发表于Circulation Research,杂志较新,但是目前的影响因子在 if = 15.2 左右,值得一读。
数据获得及方法:Illumina NextSeq 500 (Mus musculus);DCC

中枢神经系统发育过程中,神经干细胞的增殖和分化至关重要,作者通过对小鼠 NSCs 细胞分离后RNA-seq获得数据后,获得不同发育阶段的差异circRNA,并构建共表达图谱。文章发表于12月,主要为生信分析,并没有深入挖掘验证,因此影响因子并不高。
数据获得及方法:Illumina Hi-Seq 4000 sequencer 100bp PE;UROBORUS
转入正题 ==>
UROBORUS简介
Ouroboros 或 uroborus来源于古希腊,本意为一个标志,指一条咬住自己尾巴的蛇,大致如下图所示👇。而在古埃及以及罗马时代,这些代表着围绕有序世界的无形障碍,并参与了这个世界的定期更新,经常出现在一些魔法护身符。
… that the first day should make the last, that the Tail of the Snake should return into its Mouth precisely at that time, and they should wind up upon the day of their Nativity, is indeed a remarkable Coincidence, …

扯远了。。来
UROBORUS 由Perl语言编写,基于TopHat 及 Bowtie 工具,用于鉴定总RNA-seq (rRNA depleted) 的circRNA鉴定。
circRNA鉴定流程
基于大部分人类外显子circRNA的形成由经典的剪切信号指导,因此需要首先使用TopHat及Bowtie识别反向剪切的外显子中junction reads。
# 首先需要安装相应的程序
# hg19基因组、gtf文件下载及bowtie建索引
# 获取unmapped reads文件
$ tophat -p 4 -o SRR1027187_tophat_out /Users/.../ref/hg19.fa /Users/.../testData/SRR*.fq.gz /Users/.../testData/SRR*_2.fq.gz
$ samtools view unmapped.bam > unmapped.sam
UROBORUS 的主要计算步骤如下图所示
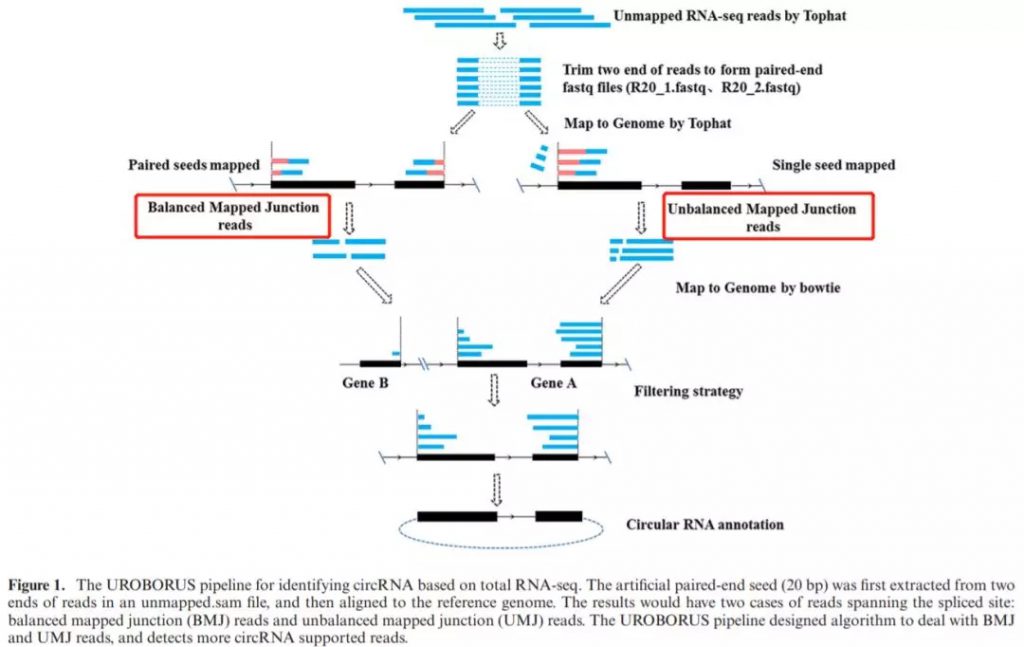
首先获取 unmapped.sam 文件中两端20bp的序列,并构建fas格式文件。然后将这个短片段比对到参考基因组 (hg19)上,最大容忍2个错配。获得mapping结果后,根据junction reads是否能完全比对两端的20bp片段,将其分为BMJ (balanced mapped junction)以及UMJ (unbalanced mapped junction)。
简单来说,BMJ 为包含两端都包含20bp的的反向剪切外显子,而UMJ 为一端中包含不满足完全20bp匹配的反向剪切外显子。并收集在3,000,000bp内且两端序列反向的候选组。对于UMJ来说,由于可能一端的片段太短不足以map到基因组上,因此使用单端匹配的时候作者过滤掉与剪切位点距离超过3bp的候选组。随后与另一端组合后用于支持circRNA的识别。
随后获得的再次使用bowtie比对到参考基因组上,过滤掉不在同一条染色体上的paired-end reads。获得超过两个以上的junction reads支持的候选circRNA。
性能比较
不难发现,UROBORUS 的鉴定原理与 find_circ 的非常的类似,在测试数据中会有什么优势呢?
假阳性
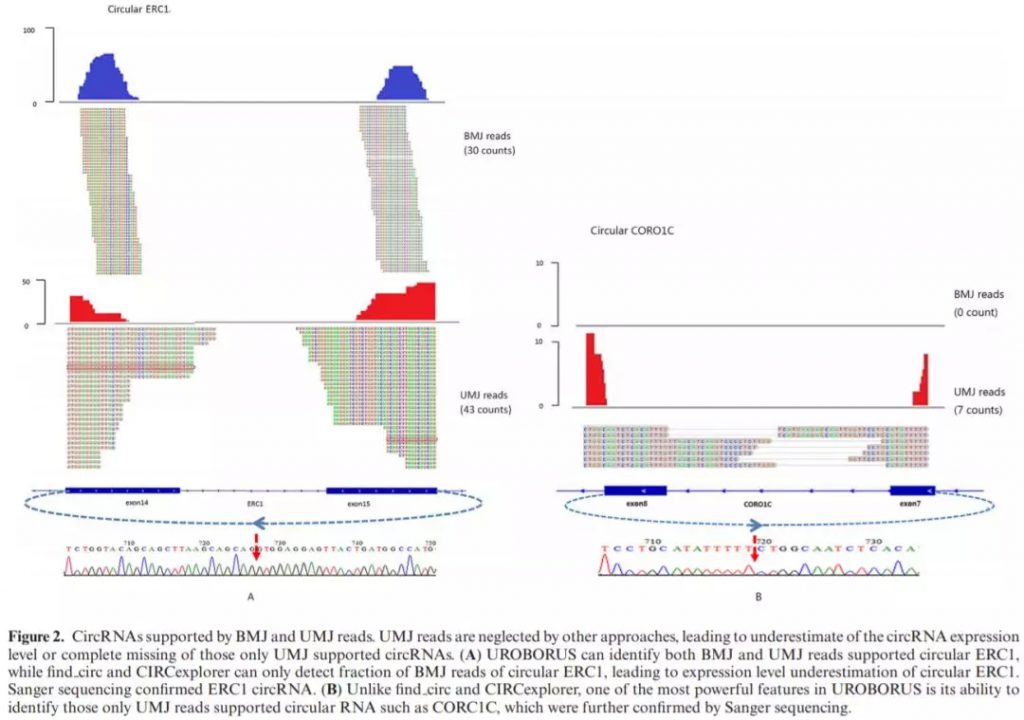
对于circRNA识别来说,少于20bp的短片段种子序列会匹配到多处参考基因组上,因此首选将片段分为两组 (BMJ & UMJ ) ,能够减少忽略这些短片段而造成的circRNA识别假阳性。从测试数据上来看,如下图所示,左边 circular ERC1序列区域包含30个 BMJ read counts,而 UMJ 有43个;而在右图 circular CORO1C中,BMJ忽略了该序列,而UMJ上能够发现有7个 read counts。上述两条circRNA序列皆由Sanger sequencing证实存在,circular ERC1能够被 find_circ及 CIRCexplorer识别,而circular CORO1C只在 UROBORUS 的鉴定结果中出现。
随后作者检测了在ploy(A)+ RNA-seq上使用UROBORUS数据集,结果显示在约1,270万个reads中总共获得2条支持的circRNA预测结果,假设全部都为假阳性结果,那么假阳性率为每百万读数约0.79。因此,作者预估UROBORUS的FDR <0.013。
多维度比较
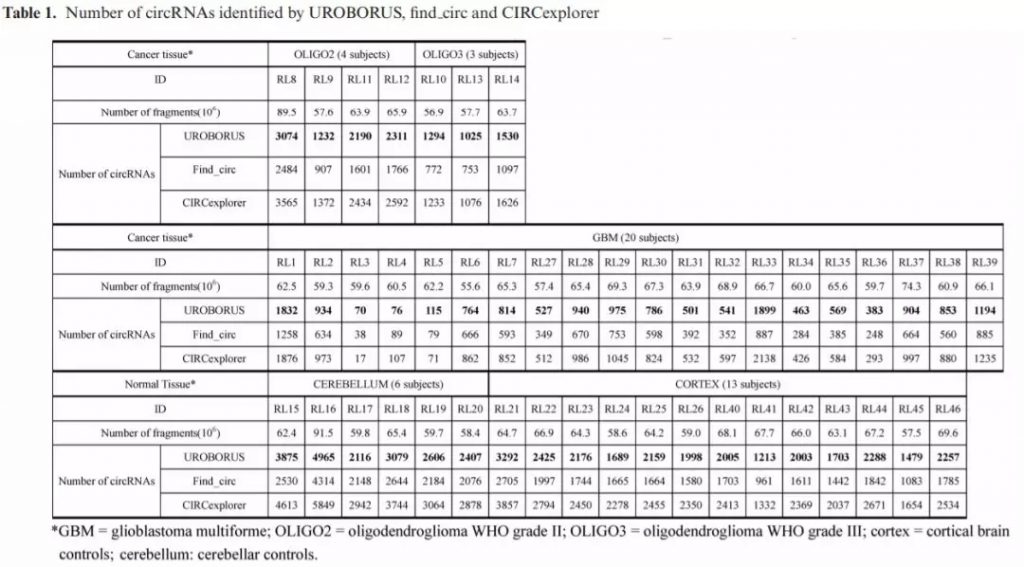
作者从不同程序鉴定的circRNA数量、表达水平、计算成本以及实验验证几个方面来比较不同程序之间的差异,如下表内容所示。
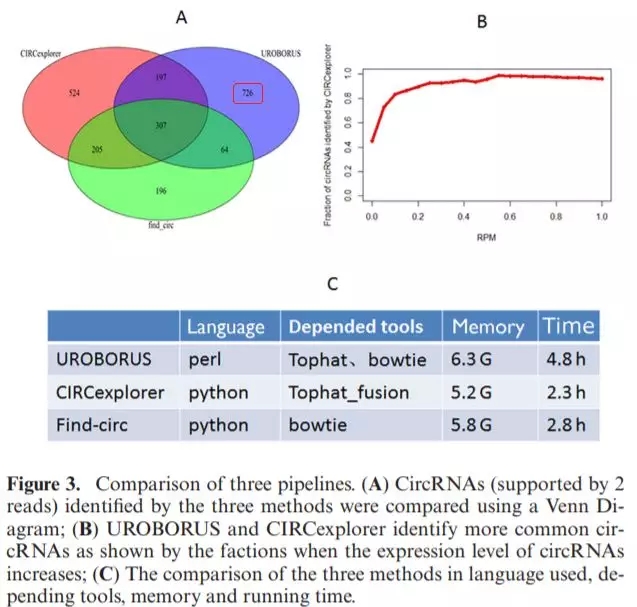
从Venn图中可以发现,三种程序全都能识别的序列为307条,其中90%的circRNA表达水平高于0.3 RPM (reads per million mapped reads) ,find_circ鉴定出196个不同于CIRCexplorer和UROBORUS的circRNA; CIRCexplorer鉴定了524个与find_circ和UROBORUS 不同的circRNA; 和UROBORUS鉴定了726个与find_circ和CIRCexplorer不同的circRNA。这些结果表明,UROBORUS能够检测到更多的新的circRNA,能够作为前两者的补充。在计算成本上来看,UROBORUS在耗内存和时间上都多于其他两种程序 (compute node: Intel Xeon CPU X5650@2.67GHz, 12 core, 80 G memory) 。
circRNA的实验验证上来看,UROBORUS特异识别的差异中低表达水平的circRNA的实验验证率为84.2% (16/19)。而进一步从正常脑组织、GBM及少突神经胶质瘤中随机选择27个circRNA,通过RT-PCR和Sanger测序以进行验证,并用 RNase RC处理证实24条circRNA的存在。
$ perl UROBORUS.pl -index /Users/leah1/find_circ/ref/hg19.fa -gtf /path/genes.gtf -fasta /path unmapped.sam accepted_hits.bam
小思考
UROBORUS开发于2016年,在多方面的表现都比较优秀(主要在人类)。在做测试数据的时候,还是有一些问题出现的,比如 tophat 多用于短片段的序列map (50-100bp),且在参数中默认使用的是bowtie2 index,参数 --coverage-search 一般在短SE测序数据中用于找寻新的剪切位点时使用,而其他情况使用 -no--coverage-search 。但是随着测序长度的增加,mapping更多地使用 HISAT2 。
【部分参考文献】
Song, Xiaofeng, et al. "Circular RNA profile in gliomas revealed by identification tool UROBORUS." Nucleic acids research 44.9 (2016): e87-e87.
Hall, Ignacio Fernando, et al. "Circ_Lrp6, a Circular RNA Enriched in Vascular Smooth Muscle Cells, Acts as a Sponge Regulating miRNA-145 Function." Circulation Research(2018).
Yang, Qichang, et al. "Circular RNA expression profiles during the differentiation of mouse neural stem cells." BMC Systems Biology 12.8 (2018): 128.

来第一个抢占沙发评论吧!