







The pervasive expression of circular RNAs (circRNAs) is a recently discovered feature of gene expression in highly diverged eukaryotes. Numerous algorithms that are used to detect genome-wide circRNA expression from RNA sequencing (RNA-seq) data have been developed in the past few years, but there is little overlap in their predictions and no clear gold-standard method to assess the accuracy of these algorithms. Researchers from Stanford University School of Medicine review sources of experimental and bioinformatic biases that complicate the accurate discovery of circRNAs and discuss statistical approaches to address these biases.
- In 2012, genome-wide statistical analysis of splicing led to the discovery of the global expression of circular RNA (circRNA) in eukaryotes and found that, in hundreds of human genes, circRNA constitutes the major isoform. circRNA expression was previously overlooked owing to a combination of biases in library preparation and heuristic filters imposed by algorithms to detect unannotated splicing events.
- Assigning reads to the correct splice junction is complicated by experimental artefacts, sequence homology and degenerate sequences at exon boundaries. Even accurate assignment to annotated splice junctions, a seemingly straightforward task compared with identifying unannotated splice events, has not been solved.
- Common RNA sequencing (RNA-seq) protocols introduce technical artefacts that can appear to be putative novel splice events, including circRNA. Statistical approaches can be used to test for these artefacts to avoid high false-positive rates, without the reduced sensitivity that comes with applying stringent bioinformatic filters.
- Read count is an unreliable metric when assessing whether a splice junction is truly expressed. Statistical approaches that reduce reliance on read count have improved the accuracy of novel linear splice detection, enabled the discovery of circRNAs spliced by the U12 (minor) spliceosome, and reduced false-positive circRNA owing to highly expressed homologous genes.
- There is little overlap in the predictions between published circRNA detection algorithms, and the field lacks a clear gold standard for assessing the accuracy of their genome-wide predictions. RNase R resistance is useful for validating a predicted circRNA, but more work is needed on normalization and appropriate enrichment tests for RNase R to be useful for assessing genome-wide accuracy.
- The ubiquitous expression of circRNA, as well as high circRNA expression from specific genes, is conserved across highly diverged eukaryotes. Conservation, as well as evidence of tissue- or development-specific regulation, provides circumstantial evidence that circRNAs are functional, although the function of most remains unknown.
Challenges for circRNA detection in RNA-seq
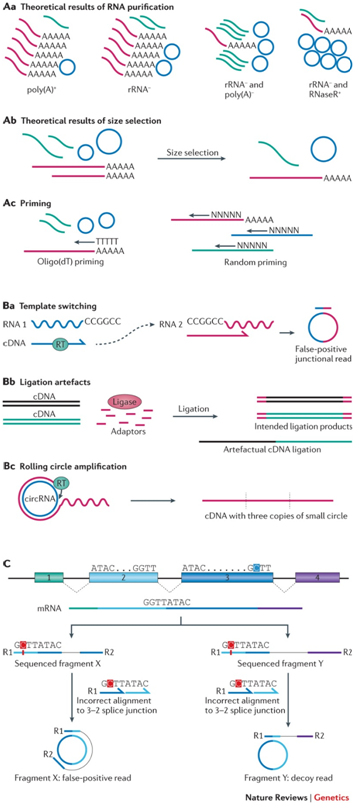
Aa–Ac | Variations in preparation protocols alter the amount of circular RNA (circRNA) in a library. Poly(A) RNA is shown in pink, non-poly(A) RNA is shown in green and circular RNA is shown in blue. Aa | Common RNA purification methods, in order of increasing relative amounts of circRNA. circRNAs are depleted by poly(A) selection and retained in ribosomal RNA (rRNA)− libraries. They constitute a large proportion of reads in an rRNA− library that has also been depleted of poly(A) RNA, and are the primary RNA in RNase R-treated libraries. Ab | Size selection excludes very small circular and linear RNA. Ac | Oligo(dT) priming biases against circRNA. Ba–Bc | Known sources of artefacts from common RNA-seq protocols. Ba | Reverse transcriptase (RT) can join two distinct RNA molecules in a non-canonical order, particularly when the two RNAs contain a common sequence. Bb | Two distinct cDNAs may be ligated together in non-canonical order during adaptor ligation. Bc | RT can displace cDNA from the template, generating a single cDNA that contains multiple copies of a circRNA. C | A convolution of homology and sequencing errors can lead to false alignments to a backsplice junction. In this case two fragments generated from a linear exon 2–exon 3 splice junction are sequenced with an error and incorrectly aligned to an exon 3–exon 2 backsplice. If the mate aligns outside the genomic region defined by the backsplice junction it is correctly discarded as a false positive, but if the mate aligns within the presumed circle it is incorrectly considered evidence of circRNA. For clarity, the mRNA sequence shown is the DNA equivalent.
Szabo L, Salzman J. (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet 17(11):679-692. [
来第一个抢占沙发评论吧!