







Abstract
CircRNAs are novel members of the non-coding RNA family. For several decades circRNAs have been known to exist, however only recently the widespread abundance has become appreciated. Annotation of circRNAs depends on sequencing reads spanning the backsplice junction and therefore map as non-linear reads in the genome. Several pipelines have been developed to specifically identify these non-linear reads and consequently predict the landscape of circRNAs based on deep sequencing datasets. Here, we use common RNAseq datasets to scrutinize and compare the output from five different algorithms; circRNA_finder, find_circ, CIRCexplorer, CIRI, and MapSplice and evaluate the levels of bona fide and false positive circRNAs based on RNase R resistance. By this approach, we observe surprisingly dramatic differences between the algorithms specifically regarding the highly expressed circRNAs and the circRNAs derived from proximal splice sites. Collectively, this study emphasizes that circRNA annotation should be handled with care and that several algorithms should ideally be combined to achieve reliable predictions.
INTRODUCTION
Long non-coding RNAs (lncRNAs) belong to a diverse class of transcripts whose common feature is that they are predicted not to function as messengers for protein translation. Instead, lncRNAs typically function as regulators of protein coding gene expression. The modulation mediated by lncRNAs can take place at every step in the gene expression pathway from transcription and chromatin remodelling to translation as well as through regulation of resulting protein function involving a wide range of different mechanisms. The mechanisms discovered to date span from lncRNAs serving as guides for proteins to lncRNAs that act as molecular scaffolds with gene regulatory proporties, thereby facilitating formation of active regulatory complexes. Additionally, lncRNAs can act as target decoys by redirecting binding of either microRNAs (miRNAs) or DNA-/RNA-binding proteins from the intended target as well as bind to and allosterically modifying the function of regulatory proteins (1). Hence, lncRNAs contribute to correct and timely regulation of protein expression and are essential for the survival and maintenance of diverse cell functions.
Circular RNA (circRNA) constitutes a particular intriguing class of recently recognized lncRNAs. Although the presence of circRNAs in human cells was established more than twenty years ago (2–5), the prevalence and abundance of these circular RNAs in human cells has only recently been revealed (6–8). Since many large-scale RNA sequencing applications rely on accessible termini or poly(A)-tail purification steps, circRNAs have evaded recognition or simply been discarded as artefacts during standard processing, which involves alignment to the ‘linear’ genome (9). circRNA are all characterized by a non-linear ‘backsplicing’ event between a splice donor (SD) and an upstream splice acceptor (SA) in contrast to a downstream SA in conventional linear splicing. Hence, elucidation of circRNA abundance requires application of dedicated bioinformatic pipelines directed to search specifically for circRNAs in datasets generated from deep-sequencing of eukaryotic rRNA-depleted RNA (6–8,10–12). These pipelines all identify circRNAs based on the presence of backsplice junction-spanning reads. As a consequence, large numbers of circRNAs derived mainly from exonic regions, but also from intronic, intergenic and UTR regions, lncRNA loci and antisense to known transcripts were identified (6,7). These analyses also revealed that multiple circRNAs may arise from the same gene locus, a phenomenon termed alternative circularization (3,6,8,10) and that circRNAs may comprise single to multiple exons (10). Although the number of circRNAs identified vary widely from >25 000 in one study (6) to a few thousands in others (7,8), it has become clear that circRNA constitutes an abundant and fascinating class of lncRNA. While most circRNAs are modestly expressed in cells, specific circRNA species are highly abundant (8) including the CDR1as/cirRS-7, which is highly and widely expressed in the brain (13). Aside from CDR1as/ciRS-7, which acts as a miR-7 sponge (7,14) and circMbl that acts as a decoy for its own protein product muscleblind (15), not much is currently known regarding the functional importance of circRNA.
A repository of circRNA has been developed, termed circBase (16), containing all annotation information on circRNAs predicted and identified thus far. To ensure that the circBase repository only describes bona fide circular RNAs, it is important that the prediction algorithms used to identify circRNAs are stringent and reliable. Here, we compare the output from 5 different circRNA prediction algorithms and evaluate the circular nature of each predicted species by its resistance to RNase R treatment known to specifically enrich for circRNA (6). Our results show that the 5 prediction algorithms yield highly divergent results and a high level of false positives, underscoring the need for further validation for example by using multiple prediction algorithms or ideally include a sequencing library of RNase R treated RNA.
MATERIALS AND METHODS
Prediction of circRNA
RNAseq libraries were downloaded from the Sequence Reads Archive (accession SRR444655, SRR444974, SRR444975, and SRR445016). The human genome was downloaded from UCSC Genome Browser (hg19) and indexed with default parameters using Bowtie1 (v0.12.8), Bowtie2 (v2.2.3), STAR (v2.4.0h) and BWA (v0.7.5a). For each RNAseq library, circRNA prediction was performed with five different algorithms (see Table 1) adhering to the suggested settings by the respective authors. Gene-annotations, required for CIRCexplorer and MapSplice, were collected from UCSC genome browser (UCSC Genes track) and Ensembl (Homo_sapiens.GRCh37.66.gtf). The predictions were executed on two Intel Sandy Bridge E5–2670 CPUs with 64 GB memory except for CIRI where 128 GB memory was allocated.
Analysis of prediction
For each algorithm, the prediction output from all four libraries was merged with custom python scripts (available upon request) into BED files (see supplementary zip-file). These files were then processed and analysed using R (see script.R in supplementary zip-file) where, for each algorithm, only circRNAs with at least three reads in one of the control samples were kept for analysis. The average count of reads spanning the backsplice junction in the two control samples was used as a measure of expression level. For each algorithm, RNAse R resistant circRNAs were classified independently as entities with at least a 5-fold expression increase in RNAse R treated samples. For CIRCexplorer, we discarded the circular intronic RNA (ciRNA) candidates in the analysis. Furthermore, to enable comparison between algorithms, the starting coordinate was converted from 1-based to 0-based for circRNA_finder, CIRI and MapSplice.
Annotation of prediction
To annotate the origin of circRNAs, we determined whether the start- and end-coordinates correspond to annotated splice sites according to gene annotation from UCSC, which indicate exonic origin. If not, the start-coordinate of circRNAs was matched against annotated 5′ splice sites allowing one nucleotide offset, indicative of lariat origin. If no match was found, the circRNA was termed ‘Unannotated’.
RESULTS
Several different tools have been developed for identification of circRNAs based on high-throughput RNA sequencing (RNAseq) datasets. We set out to scrutinize and compare the performance of these different pipelines. Using the RNAseq dataset produced by the Sharpless lab (6) consisting of two non-treated RNA samples derived from Hs68 fibroblast (SRR444655 and SRR444975) and two RNase R treated samples (SRR444974 and SRR445016), we compared five different circRNA predicting algorithms: find_circ (7), MapSplice (17), CIRCexplorer (10), circRNA_finder (11), and CIRI (12). We focused the prediction on the control samples (without RNase R treatment) and used the treated samples to demarcate true circles versus artefacts based on the level of RNase R enrichment. From the control samples, the algorithms predicted between ∼1500 (circRNA_finder) and ∼4000 (CIRI) different circRNAs with at least three reads spanning the backsplice junction identifying a total of 5075 unique circRNAs identified. Of these, only a modest overlap of 854 circRNAs (16.8%) was observed between all five algorithms (Figure 1A), indicating that the obtained circRNA landscape differs quite dramatically depending on the algorithm of choice.
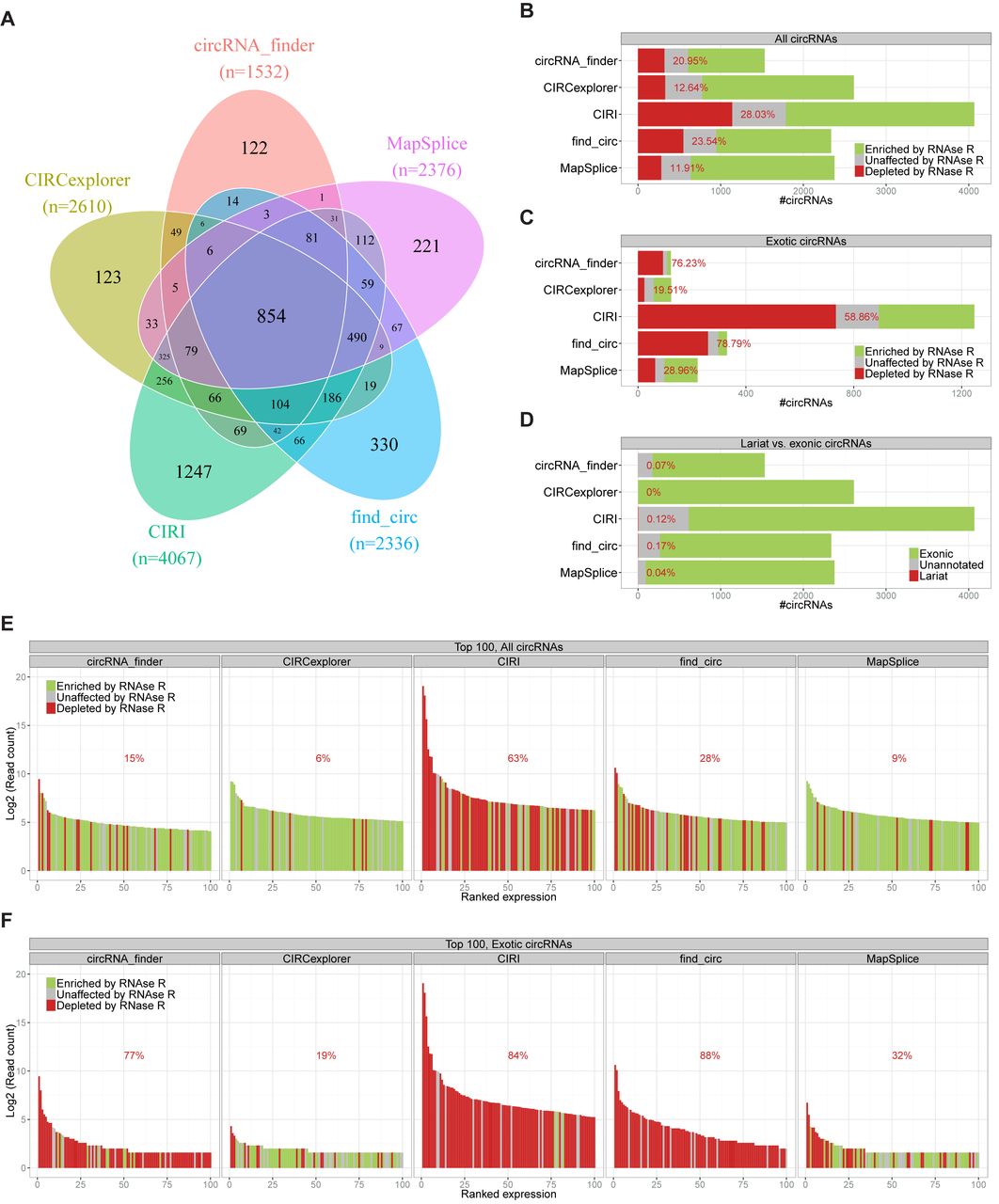
Figure 1.
Prediction of circRNAs by five different prediction algorithms. (A) Venn diagram depicting the overlap between the five different circRNA prediction algorithms. (B and C) Stacked barplot of RNase R resistance of the all predicted circRNAs (B) or exotic circRNA (C, only found by one algorithm) divided into RNAse R resistant (green), Unaffected (grey) and RNAse R sensitive (red), as denoted. Percentage reflects the fraction of RNAse R sensitive circRNAs. (D) Stacked barplot of circRNA annotation divided into exonic (green), unannotated (grey), or lariat (red). (E and F) Ranked plot of the top 100 expressed circRNAs (E) or top 100 exotic circRNAs (F) predicted by each algorithm color-coded as in B. Percentage reflects the fraction of RNase R sensitive circRNAs (false positives) within the plotted top 100.
To assess the level of false positive circRNAs, we compared the number of circRNA-supporting reads obtained in the RNase R digested samples compared to control. A 5-fold increase in read-count was defined as significant enrichment, while reduced or intermediate read-count was defined as depleted or unchanged, respectively. The algorithms exhibit between ∼12% (MapSplice and CIRCexplorer) and ∼28% (CIRI) mis-annotation of circRNAs based on the above definition (Figure 1B).
Of all the circRNAs predicted, 2043 (∼40%) were only found by a single algorithm. These constitute an ‘exotic’ group of circRNA that would be missed if the prediction was not performed by the particular algorithm. We analysed this subset of circRNAs in terms of RNase R resistance (Figure 1C). Here, except those found by CIRCexplorer and MapSplice, more than half (ranging from 59 to 79%) of the circRNAs were RNase R sensitive and thus defined as false positives, suggesting that in most cases, circRNAs not picked up by several algorithms are likely to be artefacts. Focusing on the resistant circRNAs, a total of 3251 different species were predicted by combining the algorithms, and here only 603 circRNAs (18.5%) were identified by all five algorithms (Supplementary Figure S1A), which is a small increase compared to the complete list of circRNAs. However, here ∼22% of RNAse R resistant circRNAs were identified as part of the exotic group compared to 40% above, emphasizing that exotic circRNAs in general are less likely to be true positives.
Apart from circRNAs, lariats are also resistant towards RNase R and produce non-linear reads spanning the 5′-2′ branchpoint junction. Thus, by the above demarcation, lariats would be indistinguishable from circRNAs, and even though lariats in general are highly unstable in contrast to circRNAs, specific non-debranched lariats have been shown to exhibit long half-lives (18). Therefore, based on established gene annotation, we examined the number of circRNAs derived from exonic regions or from intron regions as expected for lariats. Here, only a very small proportion (<2%) of circRNAs were of the lariat-type (Figure 1D) and we conclude that all algorithms distinguish lariats from exonic circRNAs very efficiently.
The above analyses are based on all predicted circRNAs. Of particular interest are often the most highly expressed circRNA candidates and thus the expression level should be considered when assessing the quality of each circRNA. Therefore, we focused on the top 100 most highly expressed candidates in the five different prediction outputs and evaluated the RNase R resistance of each candidate (Figure 1E). Here, the algorithms differ dramatically: 63% of the top 100 candidates predicted by CIRI do not qualify as circRNAs and none of the top 10 species are RNase R resistant (i.e. 5-fold enriched upon RNase R treatment). Also, the top 100 in find_circ prediction performs worse compared to the bulk output, suggesting that find_circ and CIRI are ‘distracted’ by highly expressed linear RNA species, and possibly less stringent regarding the mapping quality of individual sequencing reads. Focusing specifically on the exotic and uniquely predicted species, only CIRCexplorer outputs reliable circRNAs in the top 100 fraction (Figure 1F). MapSplice also performs adequately, but here most of the top 10 expressed circRNAs are not bona fide circles. In contrast, the vast majority (75–88%) of exotic circRNAs predicted by circRNA_finder, CIRI or find_circ are depleted by RNase R, and therefore exotic circRNAs found by these algorithms are at large mis-annotations.
To assess the sensitivity of the predictions, we turned to the common 854 circRNAs found by all algorithms. Based on the number of reads assigned to each circRNA by the individual prediction algorithms, CIRI and circRNA_finder exhibit the highest and lowest level of sensitivity, i.e. number of reads per circRNA, respectively (22 versus 9 reads per circRNA on average, Figure 2A and B). This is also in part reflected in the total number of circRNAs predicted, where find_circ and circRNA_finder output the lowest number of circRNA species (2336 and 1532, respectively) compared to MapSplice, CIRCexplorer and CIRI (2376, 2610 and 4067 species, respectively, see Figure 1A). Inherently, based on the complete catalogue of 3251 RNAse R resistant circRNA species, the false negative rate defined as circRNAs missed by each algorithm was inversely related to the sensitivity (Supplementary Figure S1B and C). It is also evident that the increase in sensitivity (i.e. true positives) observed for CIRI comes with a specificity trade-off (Supplementary Figure S1D).
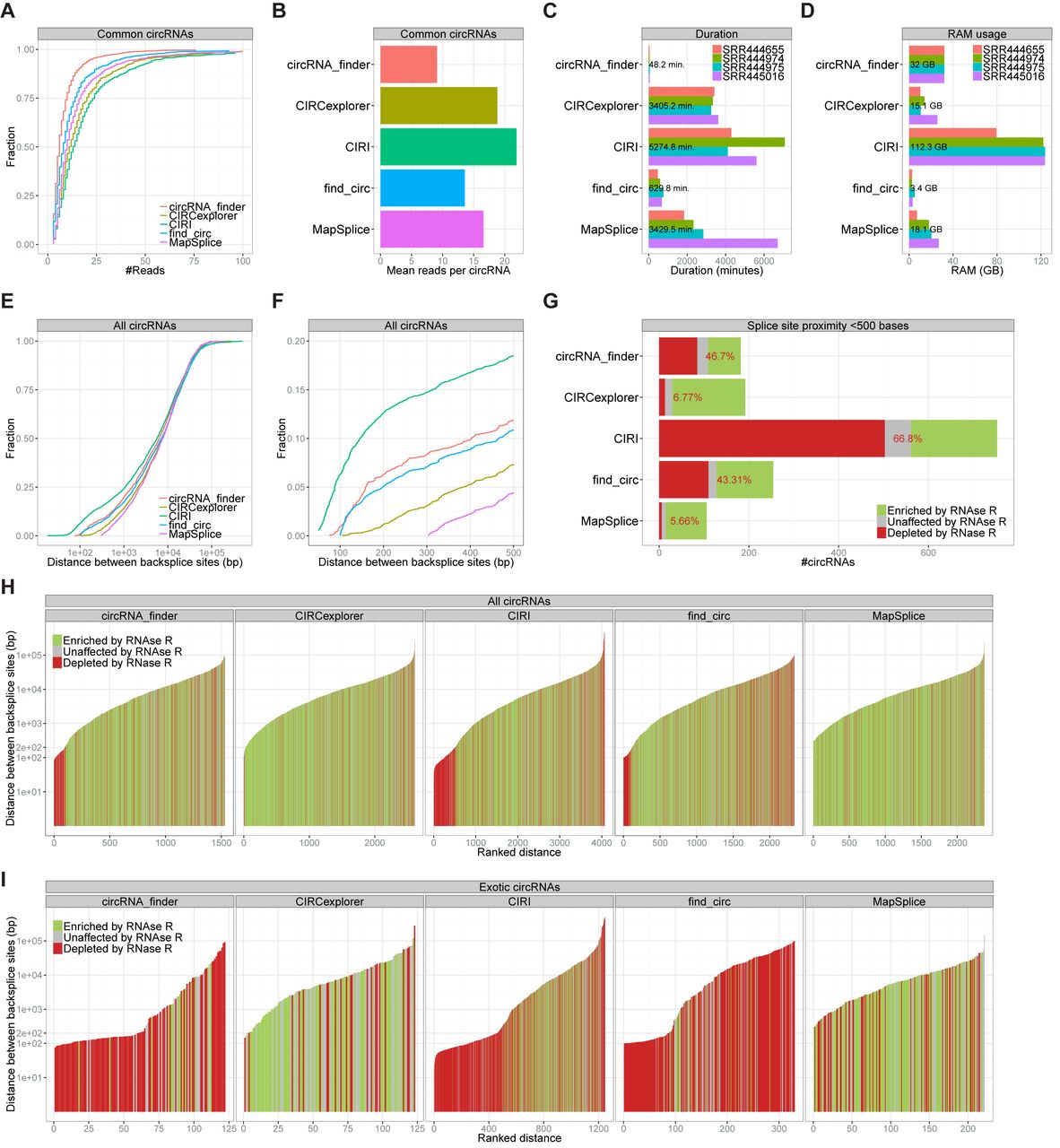
Figure 2.
Sensitivity and splice site distance. (A and B) Cumulative plot of readcount (A) and barplot showing mean number of reads (B) for the 854 circRNA species predicted by all five algorithms. (C and D) For each algorithm, the duration in minutes (C) or the max RAM usage in gigabytes (GB) (D) predicting circRNAs in datasets as denoted. Numbers reflect average duration or average RAM usage. (E) Cumulative plot of splice site distances for the circRNAs predicted by each algorithm. (F) As in E but with delimited X-axis scale. (G) Barplot as in Figure 1B of circRNAs with splice sites below 500 bp apart. (H and I) Ranked distance plot of all circRNAs predicted (H) and exotic circRNAs only (I) colorcoded as denoted.
Furthermore, sensitivity also correlates roughly with the duration of the circRNA prediction on the four datasets ranging from hours (find_circ and circRNA_finder) to several days (CIRCexplorer, CIRI and MapSplice) (Figure 2C). Apart from the duration of prediction, the RAM usage of each algorithm was dramatically diverging (Figure 2D): Only find_circ has the ability to process large datasets on standard desktops (with 8 GB RAM) using an average 3.4 GB for each dataset. Between 10 and 30 GB of RAM is required for CIRCexplorer, MapSplice and circRNA_finder, whereas CIRI demands around 120 GB to complete the datasets used.
Considering all the circRNAs identified, there is an enormous variation of distances between splice sites engaged in backsplicing, ranging typically from a few 100 nucleotides to tens or even hundreds of kilobases. Thus, we compared the distribution of distances between splice sites for the 5 algorithms (Figure 2E). Interestingly, the distribution of splice site distances differs markedly in the short-distanced range (splice sites less than 500 bases apart). CIRI and circRNA_finder predict circRNAs with very proximal splice sites (<100 bases), whereas MapSplice apparently requires at least 300 bases between splice sites to be annotated (Figure 2F) even though a minimum fusion distance of 200 was used as prediction parameter (see bash.sh in supplementary zip-file). Assessing specifically the RNase R resistance of circRNA with proximal splice sites (<500 bases), it is evident that the allowance for low proximity results in high false positive output (Figure 2G). Analyzing the RNase R resistance of all predicted circRNA for each algorithm ranked by size, suggests that it is predominantly the short distanced circRNAs that are sensitive to RNase R (Figure 2H), and almost no circRNAs <200 bases (∼10%) was here characterized as bona fide circles. This is even more evident when performing a similar analysis on the exotic circRNAs specifically, where the unique outputs from circRNA_finder, CIRI, and find_circ <200 bases show mis-annotation almost exclusively (Figure 2I).
Finally, based on the observation that exotic circRNAs (i.e. circRNAs only identified by one algorithm) in general were more likely to be false positives, we combined the algorithms in pairs and focused on the common circRNAs. For all pairs, this approach resulted in similar levels of false positives (8–12%) with the total number of circRNAs predicted ranging from ∼1000 to ∼2000 candidates (Figure 3A). Combining all five algorithms reduced the false positive fraction to ∼6% and the total number of circRNAs (as seen in Figure 1A) to 854. Plotting the top 100 expressed circRNAs from all pairs (Figure 3B) and from the combined prediction (Figure 3C), a somewhat similar output was observed with a low fraction of RNase R sensitive candidates in all cases (only 6–15% depleted by RNase R), suggesting that combining any two algorithms would greatly decrease the false positive rate and in general strengthen the output quality.
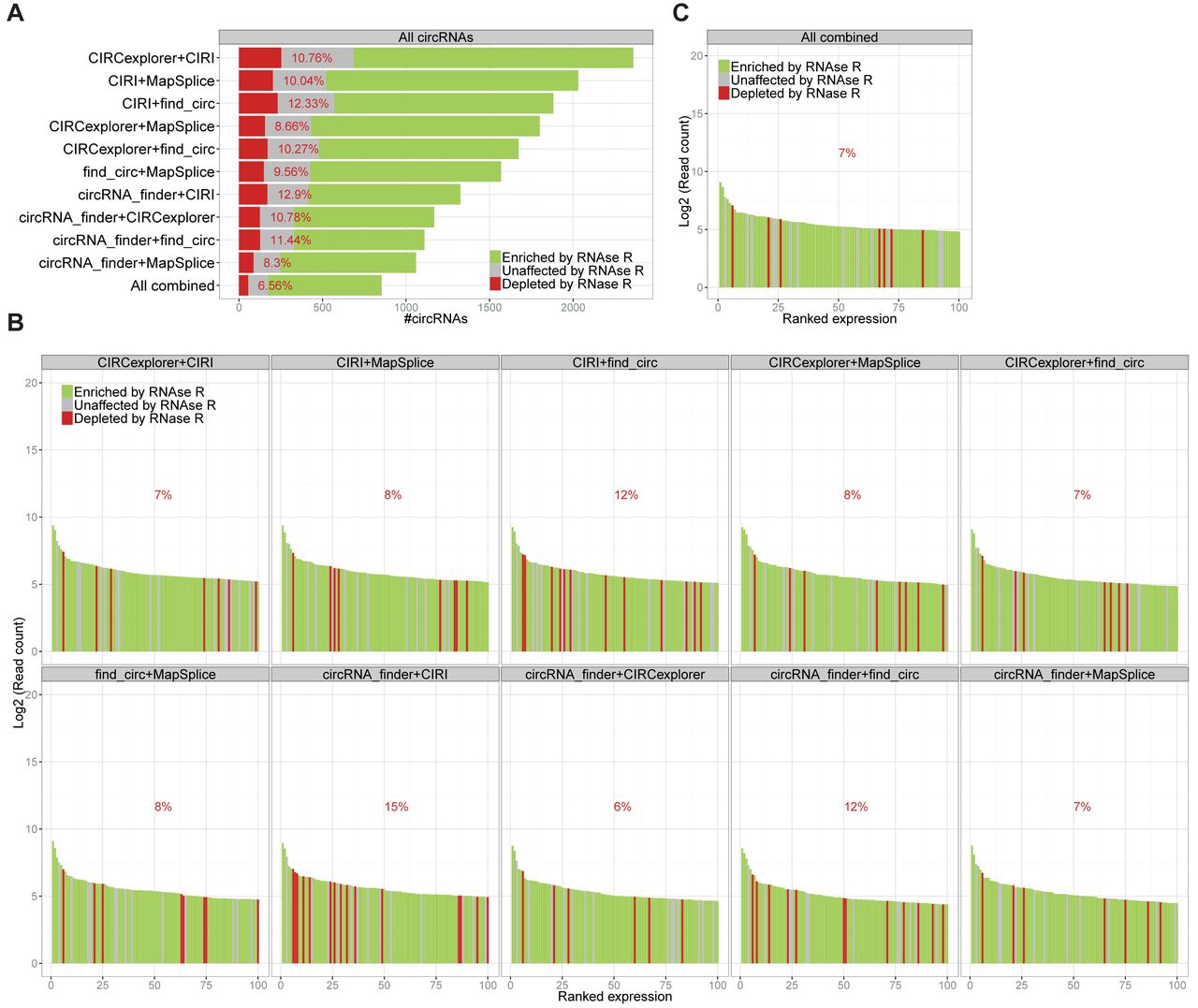
Figure 3.
Combining prediction algorithms. (A) Stacked barplot of circRNA candidates common for paired prediction using algorithms as denoted. Color coded as in Figure 1B. ‘All combined’ denotes circRNA species identified by all five algorithms. (B and C) Ranked expression plot of top 100 circRNA species identified by all algorithm pairs (B) or by all five algorithms combined (C)
as in figure 1E.
Previous Section
Next Section
DISCUSSION
Prediction of circRNA from RNA sequencing datasets is the first step towards grasping and understanding the abundance and relevance of circRNAs in cells and organisms. Characterizing the circRNome and elucidating the biology of circRNAs requires a sound and trustworthy prediction pipeline to begin with. Here, we have compared multiple aspects of five different circRNA prediction algorithms; circRNA_finder, CIRCexplorer, find_circ, CIRI, and MapSplice. Basically, the number of circRNAs predicted range from 1532 to 4067, out of which only 854 are predicted by all 5 algorithms (Figure 1A). The false positive rate measured by the RNase R resistance of circRNA candidates range from 12% to 28% (Figure 1B). Strikingly, in the fraction of highest expressed circRNAs, the false positive rate was up to 68% using CIRI (Figure 1E), which emphasizes that care should be taken to avoid mis-annotation of seemingly high abundant circular RNA. As an example, the highly abundant TUBA1B gene has high sequence similarity to the nearby and highly homologous TUBA1A gene (12). Here, multimapping reads have the potential to wrongfully ‘mix up’ conventional and non-linear splicing, which potentially explains a fraction of false positive in the circRNA output. Therefore, low map-quality reads and backsplice-junction sequences with a homology to linear exon junctions should in general be discarded or handled with great care. CIRCexplorer and MapSplice show the lowest levels of false positives, but, importantly, not at the expense of total numbers of circRNA predicted, as CIRCexplorer and MapSplice are ranked second and third regarding total number of circRNA species.
We also observe a notable difference between the algorithms concerning splice site distance requirements. Here, CIRI and circRNA_finder have the capacity to identify circRNAs with very proximal splice sites (below 100 bp, Figure 2F), however most of these circRNAs do not exhibit RNase R resistance and are therefore deemed false positives. In fact, most circRNAs below 200 bp are here classified as mis-annotated (Figure 2H and I) and therefore these species should in general be handled with care in future circRNA profiling and annotation studies.
Finally, we show that pairing up the algorithms produces a much more reliable output (Figure 3A), and therefore this approach serves as a more secure and trustworthy pipeline for circRNA annotation.
CIRCexplorer and MapSplice output the most reliable list of circRNAs, particular when addressing the exotic subset of circRNAs (Figure 1C and F) and the highly expressed circRNAs (Figure 1E), however, in our hands these two algorithms take 2–3 days to complete individual dataset predictions (Figure 2C). In addition, they both require gene annotation lists, which perhaps in part explains the low false positive rates, and furthermore, CIRCexplorer requires indexed genomes from both Bowtie1 and Bowtie2, and relies on tophat, bedtools, and samtools for mapping and processing of data, which makes CIRCexplorer one of the more complex pipelines to implement. For a faster and almost equally reliable output, we suggest combining the prediction from circRNA_finder and find_circ. However, while these are both simple pipelines, circRNA_finder is based on STAR mapping which has a memory requirement that exceeds the hardware on most laptops or personal computers, and compared to CIRCexplorer, less than half the number of total circRNAs is obtained (1110 versus 2610). Moreover, find_circ, circRNA_finder, and CIRI all work de novo without knowledge of gene annotations and exon-intron structures. Thus, these algorithms could prove more useful when conducting more unbiased circRNA analyses or in poorly annotated organisms.
To recapitulate our findings, we have constructed an overview of the algorithms listing the required third-party mapping tools, the ability to perform de novo prediction (i.e. without gene annotation requirements), the total number of output circRNAs with the percentage of bona fide species, and a brief assessment of the pros and cons for every pipeline (Table 1).
In the above comparison, KNIFE (19) and segemehl (20), two other recently developed pipelines for circRNA prediction, were not included as we failed to implement and successfully run these algorithms on the sequencing datasets used here.
Regardless of the algorithm used, there will always be false positives, and we suggest to the extent possible that novel and interesting circRNA candidates are individually validated and confirmed to minimize the fraction of erroneous annotation in the circBase circRNA repository.

来第一个抢占沙发评论吧!