







转自: Tree Breeder(微信公众号)
背景介绍
Circular RNAs(circRNAs)是近年来鉴定出的一种经由反向剪接形成的一类带有共价闭合环状结构的长链非编码RNA,circRNAs结构特殊,不具有5端的帽子及3’端的多聚腺苷酸的尾巴。随着研究的深入,已经在人类和其他物种中鉴定出数千种circRNAs,但是仅有少部分的circRNAs 的功能得到阐明,其中包括:作为吸附microRNAs的“海绵”、与RNA结合蛋白之间相互的竞争作用,甚至是翻译出蛋白质。由于circRNAs具有组织及发育过程特异性表达的特点,科研工作者推测circRNAs在细胞层面以及疾病的发病机理上,起到重要的作用。因此,解析circRNAs的表达特征能够进一步解释复杂性状以及疾病的基本原理。
然而,由于circRNAs缺乏多聚腺苷酸的尾巴,导致circRNAs的表达大多被忽略(传统的转录组测序技术采用polyA富集的方法,导致circRNAs无法被检测到)。本文作者认为,虽然部分研究筛选出了部分circRNAs,也在转录水平上量化了circRNAs 的表达丰度,但是受限于较小的样本量,导致不能系统剖析circRNAs在表达水平的差异。本文作者利用CMC数据库中的基因型及表达量数据,从遗传、生物及技术因素几个方面,剖析circRNAs表达水品的差异。并且通过严格的方法,检测到超过10000个高质量的circRNAs。通过circQTL分析,作者发现circQTL SNPs富集在有患病风险的位点上。
结论
人类大脑样本筛选circRNAs
作者通过对去除rRNA的RNA测序数据的分析,系统的鉴定了circRNAs(数据来源:258个精神病患者的前额背外侧皮质样本,54个情绪障碍患者,以及277个从CMC数据库下载的数据作为对照)。首先,作者用CIRI软件从头鉴定了起源于传统的GT—AG剪切的circRNAs,作者认为CIRI软件能很好的平衡精度和敏感度,并且能够输出环化的连接数(circular junction)和环化比例(junction ratio)。作者在选取了较为宽松的标准(circular junction counts≥1)的情况下,预测到潜在的反向剪接位点的数量为:9776~63781(中值为31286),同时预测到的环化连接的数目为:30188~577672(中值为143573)。
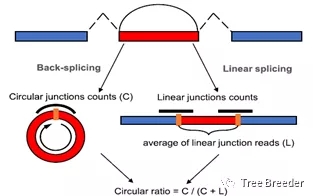
图1 circular ration示意图
此外,这些样本检测到的junction counts数,平均为基因数量的0.8%,高于其他组织核细胞系的预测结果。
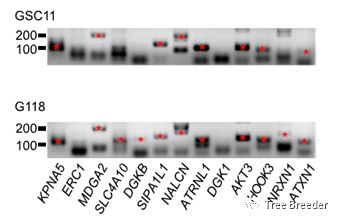
然后,作者筛选在脑组织中标表达的有潜在功能的circRNAs(筛选标准:超过半数样本中的circular junction counts≥1,平均环化率≥0.05)。最终得到10559个高质量的circRNAs,并且这些筛选出的circRNAs在至少495个样本(85%)中都有表达,其中781(7.4%)个circRNAs的表达量要高于其相对应的线性RNA。同时,10559个预测的circRNAs中,有99.7%(10526)的circRNAs能够被find_circ软件预测到。通过分析发现,预测得到的高质量circRNAs中,有大约11.6%(1229)的circRNAs是之前的研究中没有被发现的,新的种类的circRNAs。其中13种在人类的胶质瘤细胞系中检测到,其中11个通过了实验验证(运用发散引物进行了2轮RT-PCR的验证),与此同时,随机选取5个样本进行测序进行验证。结果表明这1229个新的circRNAs是可信的。
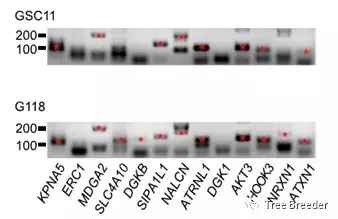
图2 RT-PCR实验结果(红星标识目标条带)
参考之前针对HEK293细胞系的circRNAs的研究,作者发现,87.1%(9198)的circRNAs位于人类脑组织样本的exon区域,7%的位于intron区域,位于基因间隔区域的则有5.9%。绝大多数(96.7%)的exonic circRNAs与编码序列重叠。预测到的9935个circRNAs来源于3916个基因,其中45.9%的基因只合成一个circRNA。为进一步探究cricRNAs的host gene的功能,作者进行了进一步的检测,作者发现alternativesplicing和splice variants显著富集在这些基因上,并且神经突触后的密集区域也检测到这些host gene 的富集,证明circRNAs在神经传导以及突触功能上有着重要的作用。
作者随后对circRNAs的表达量进行了标准化,并且评估了协变量效应。
circRNAs表达的遗传变异
为了探究遗传变异如何影响circRNAs 的表达,作者选取反向剪接位点±100kb的区域,联合高质量的circRNAs和SNPs,利用Matrix eQTL鉴定cis-sQTLs。采用q值矫正来控制FDR,作者鉴定出了和2790个circRNAs相关联的251374个circQTLs。
作者发现在circRNAs和线性RNAs的表达之间存在较弱的关联,为了排除circQTLs SNPs对基因表达的影响,作者用circular ratio代替了circCPM(circRNAs junction counts per million)进一步精细化整个分析过程。同时,作者分析发现circRNAs的表达和其host gene的表达之间存在着相互关系。
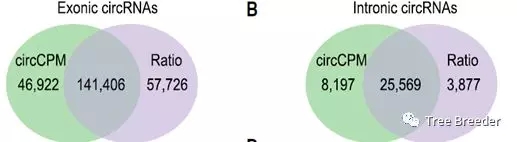
图3 鉴定出的circQTLs
CircQTL的机制
为探究circQTL的机制,作者先分析了反向剪接位点上的max-circQTLSNPs的位置分布。发现,max-circQTL更倾向于分布在反向剪接供体或者受体的附近。大约22.1%(462 of 2086)的max-circQTL SNPs位于连接位点侧翼的intron上,20.5%(427 of 2086)分布在circRNA序列上。结果表明,位于侧翼序列的SNPs更有可能参与circRNAs的调控。
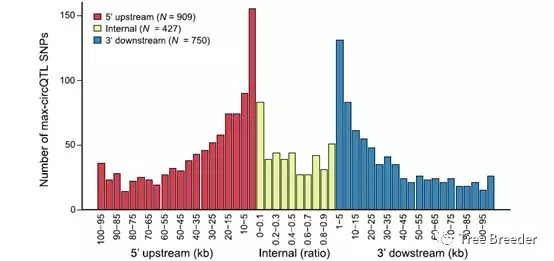
图4 circQTL SNPs的位置分布
随后,作者分析发现,在已经界定明确的元件当中,intron是circQTL数量最丰富的位置,这表明intron会影响circRNAs的环化,这与之前的研究结果相一致。早先的一个研究表明,位于intron的反向互补序列(reverse complementarysequences,RCSs)会促进circRNAs的环化。与此相对应的,作者也发现位于intron的1229个max-circQTL SNPs也存在RCSs富集的现象。
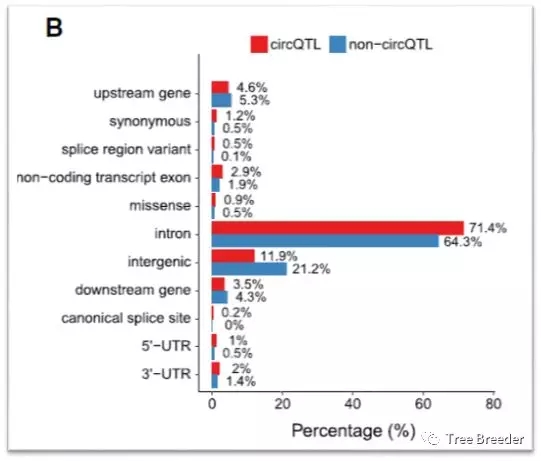
图5 circQTL分布
因为环化需要传统的反向剪接的信号,作者也检测了位于反向剪接位点的SNPs,最后发现了7个反向剪接的SNPs,并且都被鉴定为circQTL SNPs。作者发现在这7个SNPs中,canonical splice基因型的circRNAs的表达量,高于non-canonical splice基因型的circRNAs表达量,其中4类反向剪接SNPs与其circRNAs 的host gene的表达量也显著相关。作者分析结果表明,在反向剪接位点的circQTL SNPs能够通过干扰circRNAs的环化来影响circRNAs的表达。
CircQTL SNPs和疾病风险位点的关系
通过GWAS分析,发现了上千个包含疾病以及相关表型变异的基因位点。然而,表型和基因型之间的关系尚不明确。类似于其他的QTL分析,circQTL很可能帮助我们阐明遗传变异的机制。因此,作者利用GWAS数据,进行max-circQTLs的富集分析。
作者发现,相比较于non-circQTL SNPs,circQTL SNPs明显富集在和疾病相关的位点。为检测circQTL SNPs和GWAS SNPs之间高度的连锁不平衡,作者鉴定出了个122个疾病相关的157个circRNAs(一个circRNAs可能和多种疾病相关联),作者发现分别有40,35和21 种circRNAs和精神分裂症(SCZ),炎症性肠道疾病和2型糖尿病相关。有意思的是,SCZ相关联的线性RNA—AS3MT,也受到SCZ loci的的调控,意味着,线性和环状的RNA都受到SCZ位点调控的潜在机制。此外,72.6%(114 of 157)的circRNAs会受到位于侧翼intron和circRNA序列上的GWAS-linked circQTL SNPs的调控作用。这为阐明疾病的发病机理提供了新的方向。
参考文献

来第一个抢占沙发评论吧!