







转自: 生信菜鸟团(微信公众号)
上期回顾
上期我们主要提到了目前主流的circRNA鉴定程序模型分为两类:
- split-alignment-based approaches:包含 find_circ、CIRCexplorer、CIRI和 MapSplice等;
- pseudoreference-based approaches:如 KNIFE、 NCLscan等。
一般情况下,有参考注释的鉴定效果会更好,注释能够帮助算法提高对 junction read的鉴定,并且由于鉴定的候选者减少能够更好的控制错误发现率( false discovery rate, FDR)。详尽内容请翻阅 circRNA鉴定算法初窥。本期推文将介绍一款经典的程序 find_circ 的原理及实战。
find_circ简介
这是第一篇以find_circ获得人类、小鼠及线虫circRNA数据的文章,也从此来开动物circRNA研究的序幕,其中circRNA能够以miRNA sponge的方式发挥内源性ceRNA功能报道被视为circRNA功能分析中经典案例。find_circ目前已更新两版,更新内容如下:
- v1 : as used in Memczak et al. 2013
- v1.2 :
- fix the uniqueness handling. Occasionally reads would have either anchor align uniquely, but never both. These rare cases now get flagged as "NO_UNIQ_BRIDGES".
- support all chromosomes in one FASTA file
- store the size of each chromosome upon first access, pad violations of chromosome bounds by padding with 'N'
- category labels have been extended for clarity ("UNAMBIGUOUS_BP" instead of "UNAMBIGUOUS", etc.), in a manner which preserves functionality of grep commands.
- by default no longer report junctions with only one uniquely aligned anchor. Original behaviour can be restored using the --halfuniq switch.
- by default no longer report junctions that lack a read where both anchors align uniquely (NO_UNIQ_BRIDGES keyword). Original behaviour can be restored using the --report_nobridge switch
- v2 : (under development): produce multiple anchor lengths to potentially yield more junctions with unique anchor mappings.
find_circ原理
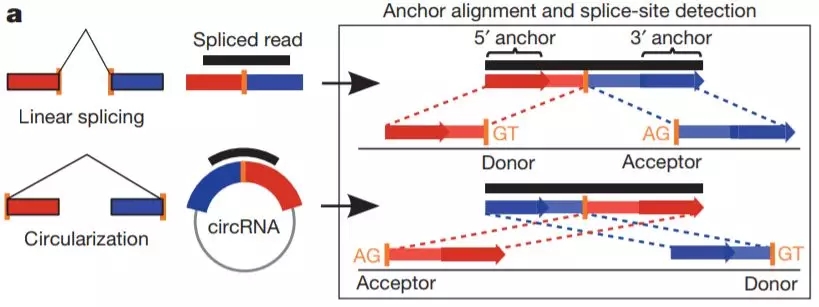
一句话来概括就是找寻反向可变剪切接头序列 (back-spliced junction) 来鉴定circRNA。
大致的步骤如下,首先获得unmapped到参考基因组上的片段,取每一个片段两端20mers分别比对到剪切的外显子上,挑取以相反方向排列的锚定位置(anchor positions),扩展对齐之间的序列,GU/AG为剪切位点。
find_circ实练
▲▲ 要注意的是分析环境,python2.7下需要安装包numpy以及pysam。
Prerequisites
- The scripts run on Ubuntu 12.04.2 on a 64Bit machine with python 2.7. We do not know if it runs in different environments but other 64Bit unix versions should run if you can get the required 3rd party software installed.
- You need to install the python packages numpy and pysam. If there are no packages in your linux distro's repositories, try the very useful python installer (building numpy requires many dependencies, so obtaining pre- compiled packages from a repository is a better option).
根据文献(文献),在NCBI上搜其对应的ftp地址,以下代码仅展示两条。
比如使用文中的数据内容(如下图)。首先下载参考基因组hg19 (905M) 并解压,并建立索引,过程需要比较久的时间所以选择放后台


$ wget http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/chromFa.tar.gz
$ tar -zxvf chromFa.tar.gz
$ cat chr*.fa >chromFa.fa
$ nohup bowtie2-bulid chromFa.fa hg19.fa &
- 下载GEO数据,由于数据并不多分条下载就可以了,或者写一个循环。R_getGEO方法详见健明团长的推送《解读GEO数据存放规律及下载,一文就够》,随后下载数据进行fastq转换。
$ wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP009/SRP009373/SRR364679/
$ fastq-dump –split-3 SRR364679.sra
$ fastqc -o qc SRR364679
- 去接头以及低质量的数据,这里使用的fastp,集去低质量去接头fastqc等过程,一套走完。
$ conda install fastp # 安装
$ source activate py27 # 这里要注意python环境,顺带把后续分析依赖的python包也一并安装
$ conda install numpy pysam
$ fastp -i SRR364679.fastq -o out.SRR364679.fq
- find_circ使用
# 下载find_circ程序包
$ git clone https://github.com/marvin-jens/find_circ.git
# 获得unmapped reads
$ bowtie2 -p16 --very-sensitive --phred64 --score-min=C,-15,0 --mm -M20 -x hg19.fa -U out.SRR364679.fq geo > bowtie2.log |samtools view -hbuS - |samtools sort - -o ./out.SRR364679.sort.bam
$ samtools view -hf 4 out.SRR364679.sort.bam | samtools view -Sb - > out.SRR364679.unmapped.bam
# 提取两端 anchors
$ python ./unmapped2anchors.py out.SRR364679.unmapped.bam | gzip > out.SRR364679.anchors.fastq.gz
# 再次比对
$ bowtie2 -p16 --reorder --mm --score-min=C,-15,0 -q -U out.SRR364679.anchors.fastq.gz -x hg19.fa | python ./find_circ.py -G chromFa.fa -p prefix -s find_circ.sites.log > out.SRR364679.find_circ.sites.bed
# 根据条件过滤筛选circRNA,包括判断方向、剪切位点信息,并删除距离超过100kb的剪切片段
$ grep CIRCULAR out.SRR364679.find_circ.sites.bed | grep -v chrM | awk '$5>=2' | grep UNAMBIGUOUS_BP | grep ANCHOR_UNIQUE | python ./maxlength.py 100000 > out.SRR364679.find_circ_candidates.bed
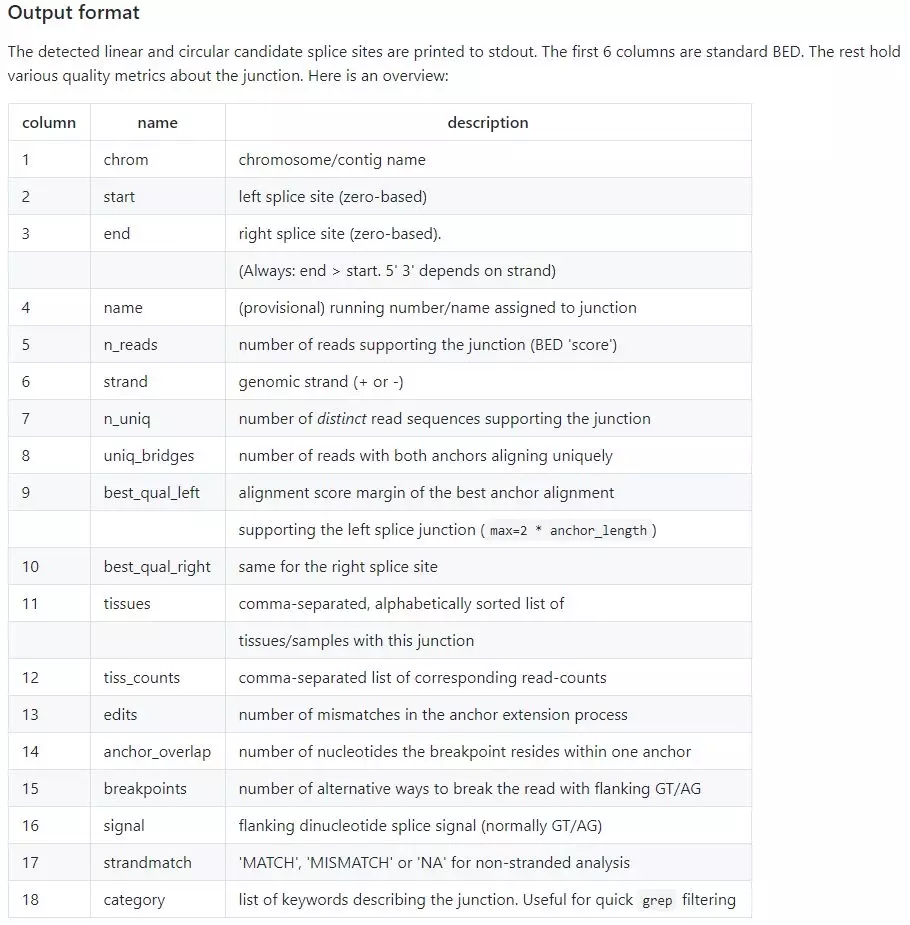
输出文件的格式如下图,可以根据关键词进行过滤筛选对应条件的circRNA_candidates,随即进入下游分析。
来第一个抢占沙发评论吧!