







转自: 生信菜鸟团(微信公众号)
背景介绍
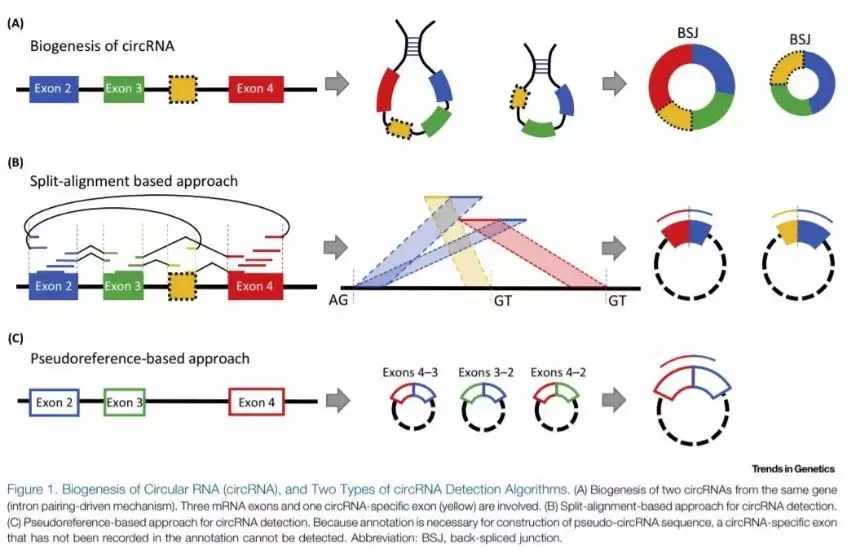
circRNA的生信分析根据鉴定的方法不同可以分为以下两种:
- split-alignment-based approaches:针对内含子驱动模式下的反向可变剪切接头序列(back-spliced junction)设计的预测软件,如 find_circ、CIRCexplorer、CIRI和 MapSplice等;
- pseudoreference-based approaches:通过基因组注释信息推测得到反向可变剪切接头序列,然后与注释的外显子序列进行匹配,预测得到新 circRNA的软件,如 KNIFE、 NCLscan等。
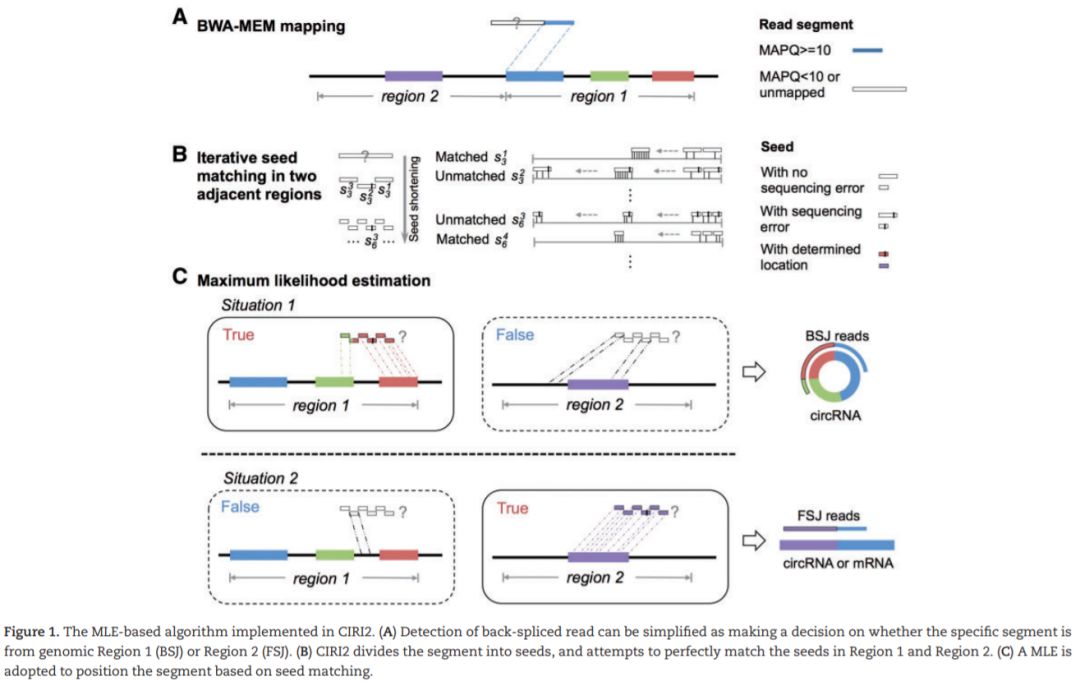
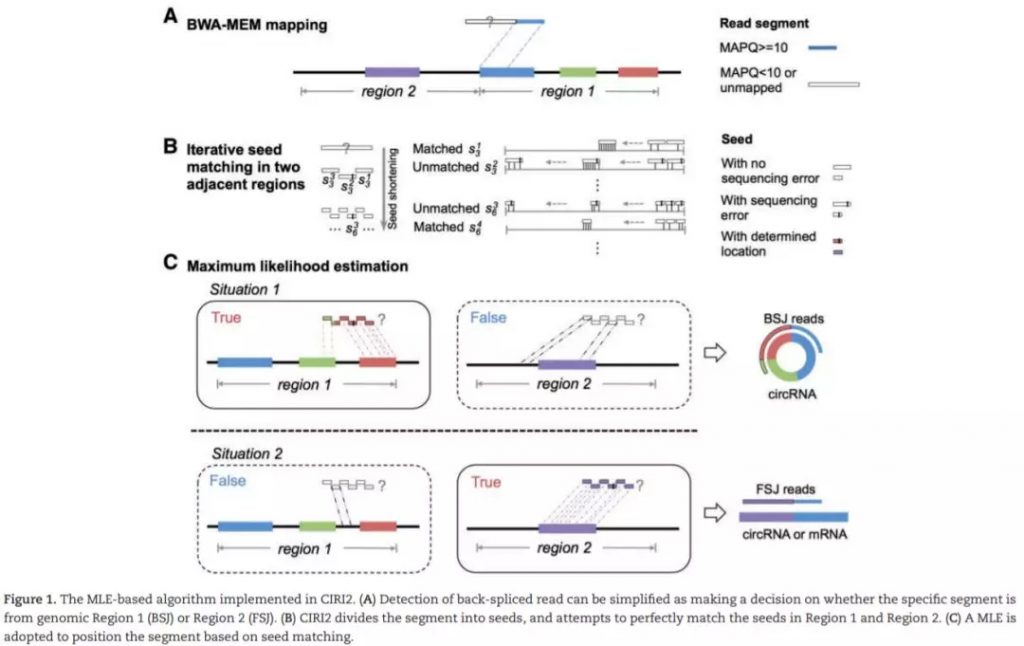
我们在前期的学习专题中,已经逐步介绍了find_circ、CIRCexplorer、KNIFE、的使用方法,今天我们就来介绍一款多线程、在鉴定BSJ过程中引入最大似然法 (maximum likelihood estimation (MLE)) 来提升鉴定效率的程序吧。
概述
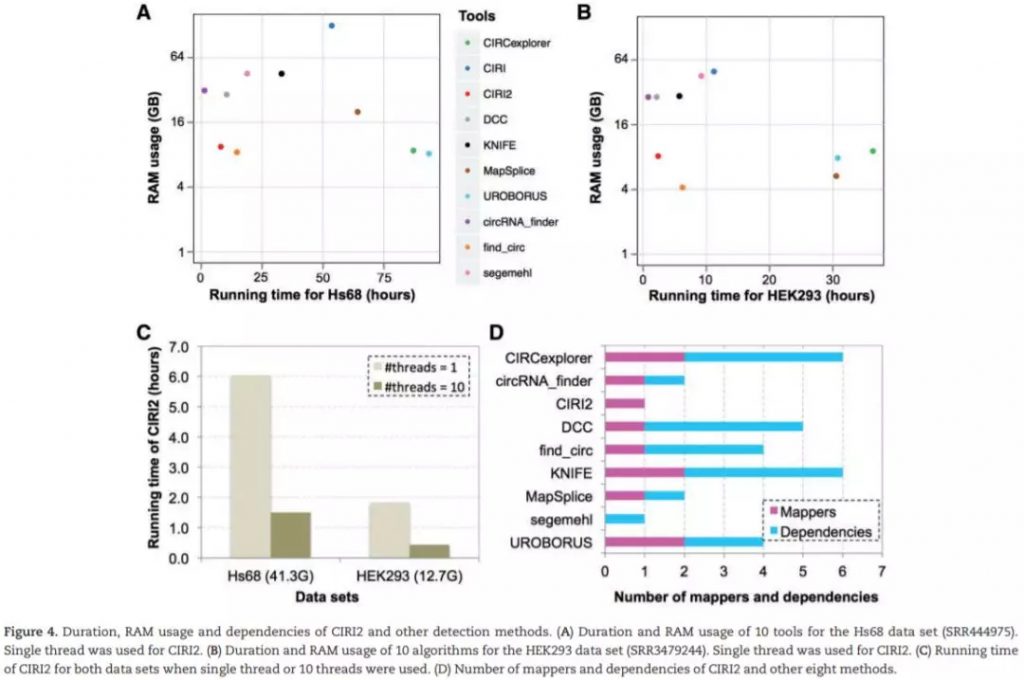
CIRI2是一款使用BWA-MEM比对结果,支持基于BSJ de novo的检测。在CIRI基础上,改进了MLE模型,判断潜在BSJ read中是否多个可能区域,有效控制由错误mapping或者基因组中重复序列所导致的假阳性。同时,CIRI2在测试数据中F1得分平均值最高,且较其他识别程序消耗更少的内存及运算时间。CIRC2需求经RNase R处理的样本数据。
( 其中 F1=2*sensitivity*(1-FDR)/(sensitivity+1- FDR) )
前期准备
$ bwa index -a bwtsw ref.fa
$ bwa mem –T 19 ref.fa reads.fq > aln-se.sam (single-end reads)
$ bwa mem –T 19 ref.fa read1.fq read2.fq > aln-pe.sam (paired-end reads)
# 对于输出SAM文件不用自行处理,CIRI2内部包含高效过滤。
使用方法
$ perl CIRI2.pl -I in.sam -O outfile -F ref.fa (-R ref_dir/)
# The arguments of CIRI2 are as followings:
-I, --in
input SAM file name (required; generated by BWA-MEM)
-O, --out
output circRNA list name (required)
-F, --ref_file
FASTA file of all reference sequences. Please make sure this file is the same one provided to BWA-MEM. Either this argument or -R/--ref-dir is required.
-R, --ref_dir
directory of reference sequence(s). Please make sure fasta files in this directory are from the FASTA file(s) provided to BWA-MEM. Either this argument or -F/--ref-file is required.
-A, --anno
input GTF/GFF3 formatted annotation file name (optional)
-G, --log
output log file name (optional)
-H, --help
show this help information
-S, --max_span
max spanning distance of circRNAs (default: 200000)
-high, --high_strigency
use high strigency: only output circRNAs supported by more than 2 distinct PCC signals (default)
-low, --low_strigency
use low strigency: only output circRNAs supported by more than 2 junction reads
-0, --no_strigency
output all circRNAs regardless junction read counts or PCC signals
-U, --mapq_uni
set threshold for mappqing quality of each segment of junction reads
(default: 10; should be within [0,30])
-E, --rel_exp
set threshold for relative expression calculated based on counts of junction reads and non-junction reads (optional: e.g. 0.1)
-M, --chrM
tell CIRI2 the ID of mitochondrion in reference file(s) (default: chrM)
-T, --thread_num
set number of threads for parallel running (default: 1)
-Q, --quiet
keep quiet when running
-D, --output_all
keep the temporary files after running (more disk space would be needed)
结果文件以\t分割,各列信息如下:
Column 1: circRNA_ID
Column 2: chromosome of a predicted circRNA
Column 3: circRNA_start
Column 4: circRNA_end
Column 5: circular junction read count of a predicted circRNA
Column 6: unique CIGAR types of a predicted circRNA. For example, a circRNAs have three junction reads: read A (80M20S, 80S20M), read B (80M20S, 80S20M), read C (40M60S, 40S30M30S, 70S30M), then its has two SM types (80S20M, 70S30M), two MS types (80M20S, 70M30S) and one SMS type (40S30M30S). Thus its SM_MS_SMS should be 2_2_1.
Column 7: non-junction read count of a predicted circRNA that mapped across the circular junction but consistent with linear RNA instead of being back-spliced
Column 8: ratio of circular junction reads calculated by 2*#junction_reads/(2*#junction_reads+#non_junction_reads). #junction_reads is multiplied by two because a junction read is generated from two ends of circular junction but only counted once while a non-junction read is from one end. It has to be mentioned that the non-junction reads are still possibly from another larger circRNA, so the junction_reads_ratio based on it may be an inaccurate estimation of relative expression of the circRNA.
Column 9: type of a circRNA according to positions of its two ends on chromosome (exon, intron or intergenic_region; only available when annotation file is provided)
Column 10: ID of the gene(s) where an exonic or intronic circRNA locates
Column 11: strand info of a predicted circRNAs (new in CIRI2)
Column 12: all of the circular junction read IDs (split by ",")
CIRI2地址:https://sourceforge.net/projects/ciri/files/CIRI2/
评估方法
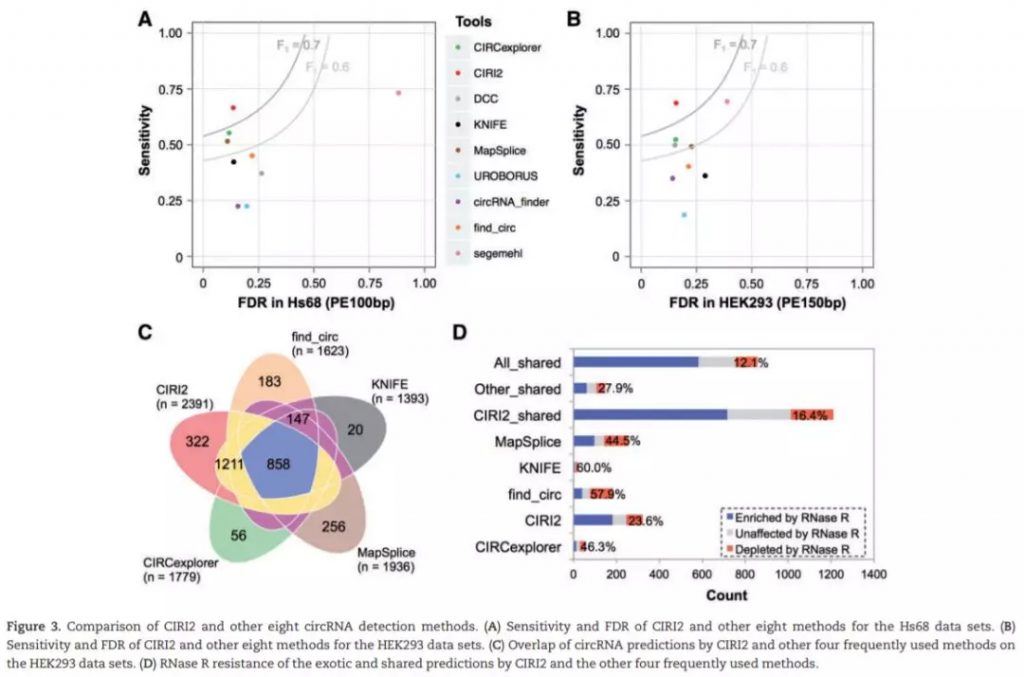
测试使用4个Hs68细胞系的数据集,包括两个RNase R处理 (SRR444974和SRR445016) 和未经RNase R处理 (SRR444655和SRR444975) ;HEK293细胞系中一个处理的SRR3479244和未处理的 SRR3479243。使用hg19基因组序列作为参考基因组。每一组数据都单独计算,并使用qstat –f Job_ID记录运行时间和RAM使用情况。对比处理和未处理组,如果未处理组中的circRNA在处理中明显富集 (at least 3-fold increase of BSJ reads count),那么认为该鉴定结果为真阳性,反之则假。FDR及sensitivity计算如下:
The ratio of false positives in all of the predictions by each tool was calculated as FDR of the algorithm.
The ratio of true positives detected by each tool in the above total circRNAs was used to evaluate the sensitivity of the tool.
检测结果如下:
命令还是很友好的,快去试一下吧 ❀。
来第一个抢占沙发评论吧!