








转自: 生信菜鸟团(微信公众号)
上期回顾
连续两期我们梳理了circRNA高通量数据获得的方式以及两者的偏好性,并提出一种提高RNA-seq分辨率的试验方法RPAD(连接)。诚然,获得良好数据的前提在于实验设计及对应的生物学意义,这里涉及到一系列专业的内容,不在此班门弄斧。但是,要提醒的是,数据至少三次生物学重复,一个样本的数据量保证在6G以上,最为稳妥。接下来的几期我们开始着手处理这些"优秀"的数据。本篇推文将对目前主流的多种鉴定方法进行初步梳理。
鉴定模型
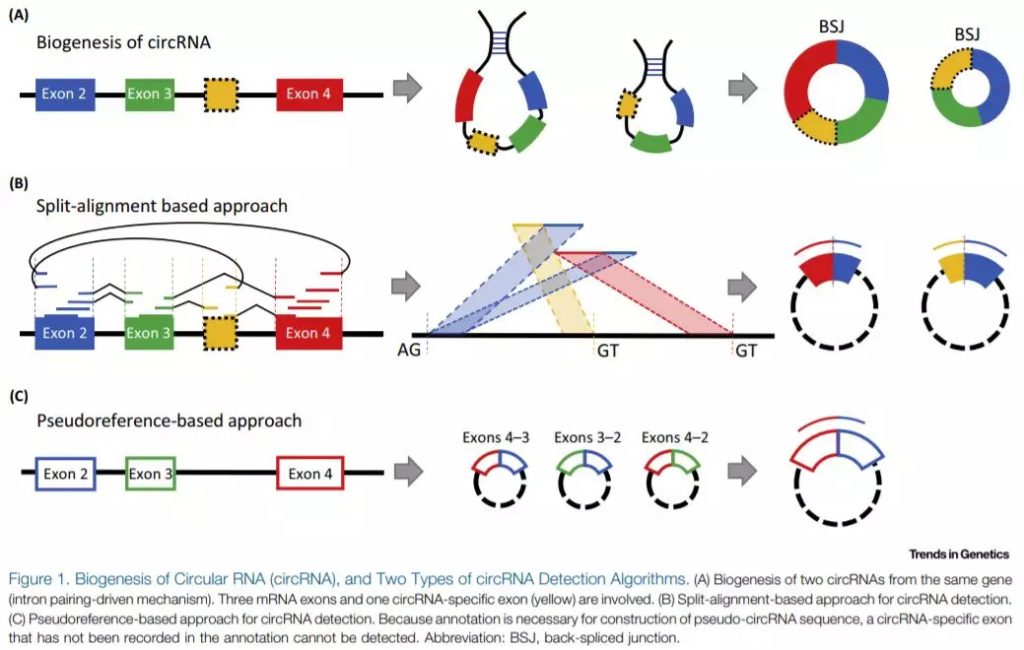
自2012年报道circRNA是一类新型的non-coding RNA开始,针对于circRNA特殊性质设计的鉴定程序已超过十种,根据不同的circRNA形成模型,可将circRNA的鉴定程序分为两大类(Figure 1):
- split-alignment-based approaches:针对内含子驱动模式下的反向可变剪切接头序列(back-spliced junction)设计的预测软件,如 find_circ、CIRCexplorer、CIRI和 MapSplice等;
- pseudoreference-based approaches:通过基因组注释信息推测得到反向可变剪切接头序列,然后与注释的外显子序列进行匹配,预测得到新 circRNA的软件,如 KNIFE、 NCLscan等。
但是,这些算法对基因组注释的依赖性与基于参考基因组的上述分类不完全相关,比如基于pseudoreference-based approaches的 NCLscan就能够在没有注释的情况进行鉴定(Table 1),而UROBORUS及CIRCexplorer则需要依赖注释来消除假阳性。虽然KNIFE起初是在外显子注释的基础上开发的,但是最新的版本中也添加了无参鉴定作为一种替补策略。相比之下,find_circ和CIRI能够完全不依赖于注释,因此它们可用于没有完整注释或只有草图基因组的生物体中的circRNA检测。
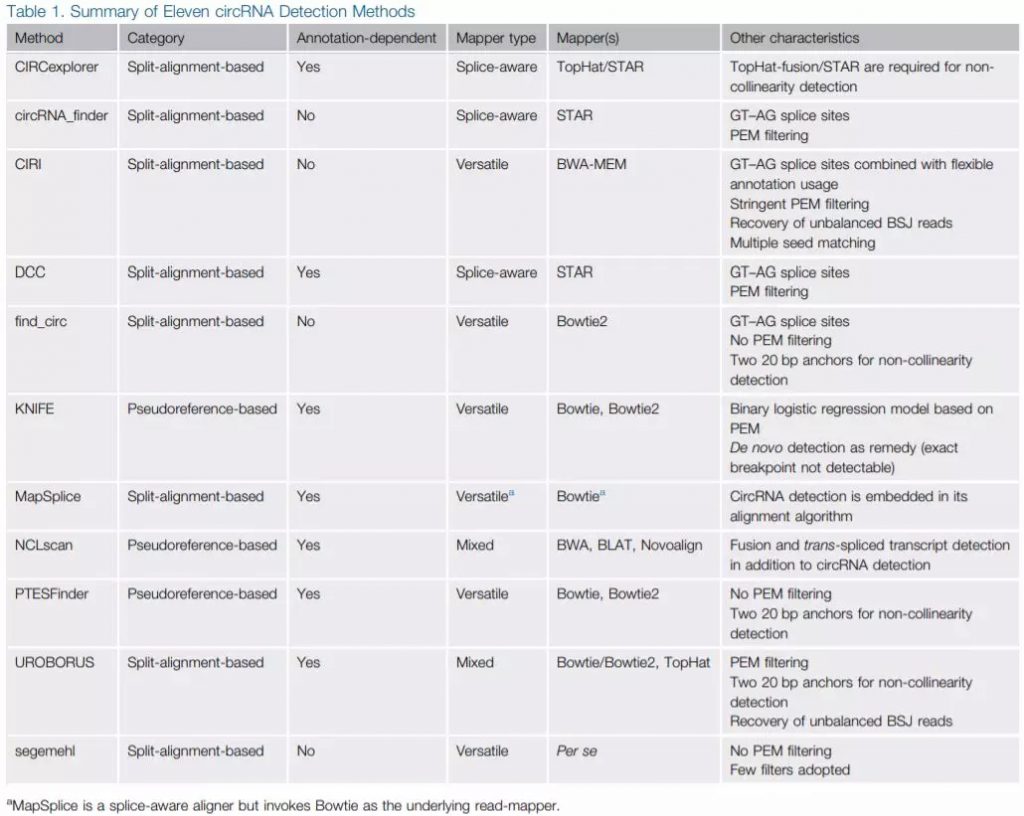
而一般情况下,有注释的鉴定会更好。目前对不同鉴定程序的评估结果显示,注释能够帮助算法提高对 junction read的鉴定,并且由于鉴定的候选者减少能够更好的控制错误发现率( false discovery rate, FDR)。
反向剪切接头读段 (BSJ reads) 的鉴定及招募
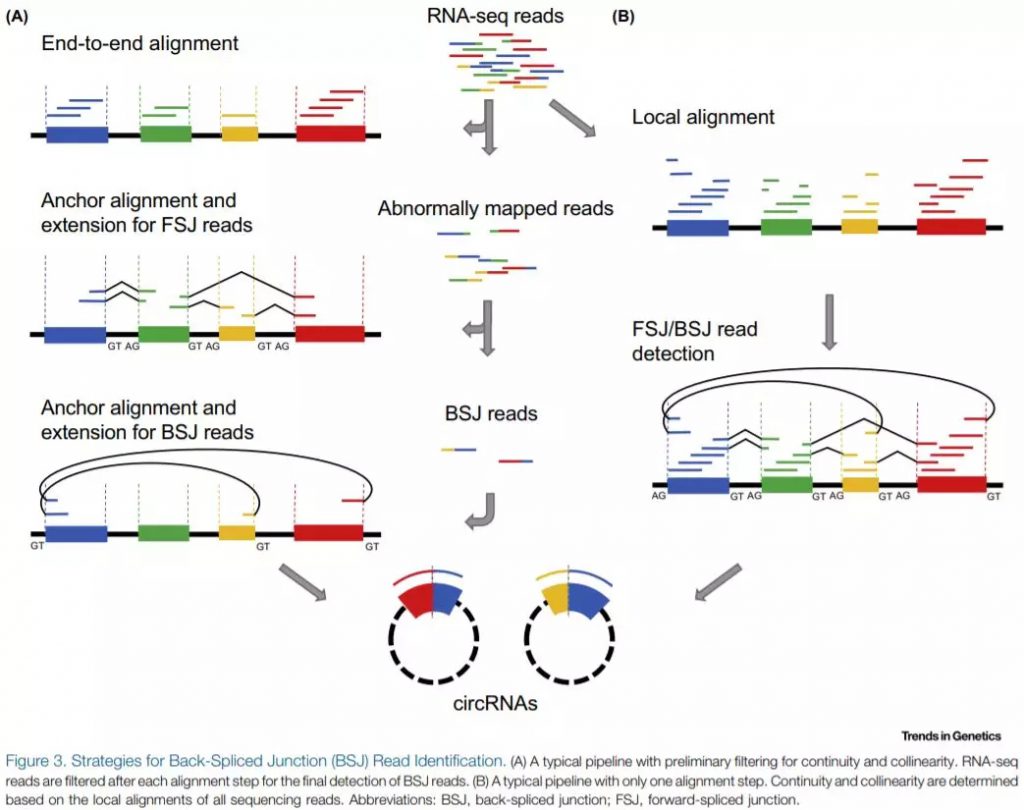
基于片段比对,不同算法的第一步都将序列比对到参考基因组。find_circ 和UROBORUS通过提取非共线的序列两端20bp单独再次比对。如果这些片段在同一条链上,且序列顺序相反,则认定其位置出现潜在的BSJ。CIRCexplorer 利用TopHat-fusion得到不同的unmpped读段(Figure 3A)。CIRI 基于所有读段和读段片段比对到参考基因组上的信息,初步过滤后,可以检测到在两个或两个以上片段的BSJ(Figure 3B)。CIRC采用最大似然法进行预测,首先根据长片段的位置确定BSJ和FSJ相对应的短片推断的基因组区域,然后将这些短片段分割成多个seed匹配到这两个区域。通过匹配计数以及相对位置的比较,可以确定该短片对最可能出现的位置。该方法提高了unbalanced BSJ鉴定的精确度,同时也保持了在低丰度是环状RNA检测的敏感度。
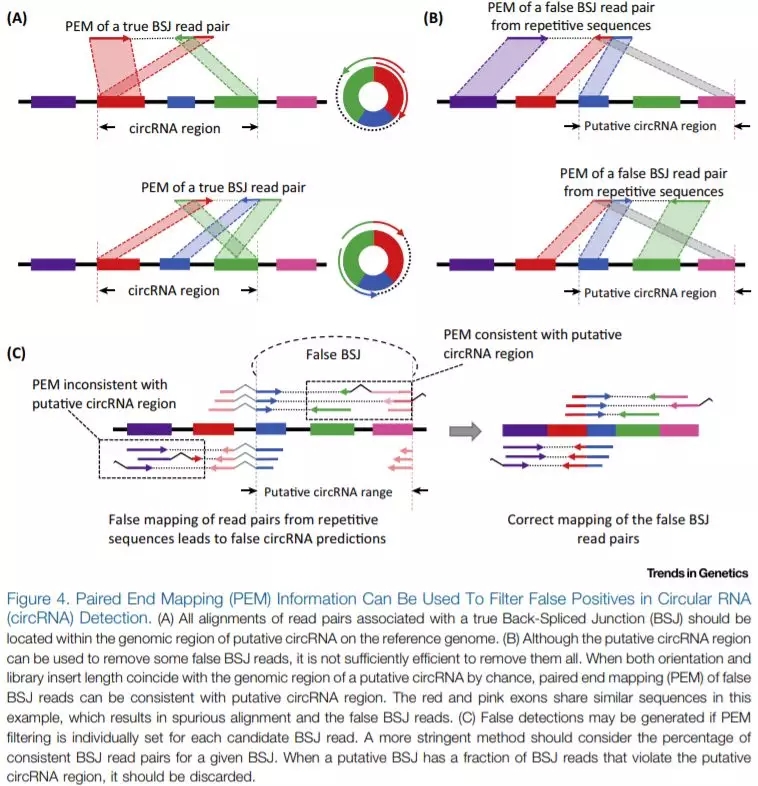
随着RNA测序组装以及比对算法的发展,双末端mapping(Paired End Mapping, PEM)将是环状RNA检测中共同的趋势,但还是存在一定的FDR,因此引出alignment score, mapping quality, and mapping length across the junctions这几个评分项目用于区分真假BSJ。注意经历复制事件的序列可能会是干扰算法主要的主要来源。通过基于在两个邻近区域内的seed匹配信息,使用最大似然法分析,可以降低同源序列在环状RNA鉴定中的干扰(Figure 4)。
小结
综上,不同的算法对于不同策略在不同侧重点上进行了优化。环状RNA的鉴定主要在于BSJ以及PEM鉴定,基于有效的参考基因组及转录本的组装和比对,以及下游分析,可以更好提高环状RNA鉴定的精度以及敏感度。
来第一个抢占沙发评论吧!