






circular RNA(circRNA)预测软件对比,circRNA的预测对于后期研究意义重大,怎么利用现有的工具去减少后期的 工作量并比提供研究中的效率显得非常重要,同时能减少成本和精力的投入,今天针对find circ、MapSplice、CIRCexplorer、circRNA finder和CIRI 五个软件进行了对比,找出了各个软件的优劣。
先看几个软件的特点:
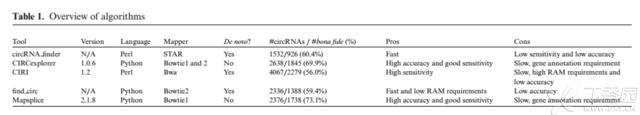
所以可以根据软件特点选择合适的软件进行应用
在网上搜到了其中三个软件的中文介绍:(为copy and paste)
"1. find_circ
这款工具是在2013年随着 Circular RNAs are a large class of animal RNAs with regulatory potency 这篇文章发布的,从而吹起了一阵研究circRNA的风潮~。
现有的所有circRNA工具的第一步都是mapping,将RNA reads mapping到基因组上,但是普通线性RNA是可以很好的mapping回去的,对于circRNA来说,成环的剪接位点不能直接mapping回基因组上。 首先,就是用比对mapping的方法筛选出这样的序列,也就是把mapping不上的序列都体取出来。 然后,将这些mapping不上的reads取两头20bp,变成短序列,重新mapping到基因组上。
这里有个参数20bp的reads,为什么要用20bp的?是否可以用更长的,或者更短的?
我们来做个简单计算。多长的一条序列可以唯一比对到基因组的某个地方? 首先,基因组的大小为3G,也就是 三十亿字节 。
,约为 十亿字节 。 那么任意取出一条15bp的read,它的碱基排列情况是4^15的其中一种,任意取出两条15bp的reads 它们完全相同的概率为。 从基因组上的任意取出15bp长的一段序列,有三十亿种可能性,取两次, 取出同一条15bp长的序列的可能性为, 这种可能性(概率)小于 任意从15bp序列的全部碱基排列情况中抽出2次相同碱基序列的概率 ,所以说15bp的read可以不唯一比对到基因组的某个地方。 同理计算16bp reads,所以任意抽出一条16bp的序列可以唯一比对到基因组上。(可能算得不对,如果有人发现错误,请立即指正,谢谢)
这样看来20bp的序列,可以唯一比对到基因组上,也可以根据测序reads的长度适当放大缩小(我觉得缩小还是没必要的,可以放大,取25bp,30bp之类的)。
接下来,将短序列mapping到基因组上后,他们开发了一个方法,检测这些短序列是否是circRNA的短序列。
需要检测的条件如下:
GU/AG 在剪接位点的两侧出现
可以检测到清晰的breakpoint
只支持2个mismatch
breakpoint不能再短序列(anchor)以里2 nucleotides 之外的地方出现,也就是最多2nt (这条可能理解有偏差)
至少有两条reads支持这个junction
mapping正确的一个短序列的位置要比它mapping到其他位置的分值高35分以上。
通过这些条件,就筛选出了文章认为正确的circRNA。
另外在这篇文章里重点介绍了circRNA的一个功能,富集microRNA,某些circRNA上有很多microRNA的seed。
2. CIRCexplorer
这款工具是在2014年随着 Complementary Sequence-Mediated Exon Circularization 的发布而问世,这篇文章我非常欣赏,我认为它是去年做RNA领域最值得读的一篇文章。
CIRCexplorer这个工具巧妙的用fusion gene这个思路去检测circRNA。 首先,过滤出Tophat无法mapping上的reads,再把这些reads用Tophat-Fusion mapping到基因组上。那些用Tophat-Fusion mapping到基因组上的非线性候选位置的reads就是潜在的back-splied juction reads。 接下来,这些reads会在有基因注释的帮助下,确定更加精确的donor, acceptor位置。 最后,对circular RNA进行注释。
这篇文章里介绍了在intron区域的 反向重复Alu序列 是引起circRNA形成的一个原因(即“内含子配对驱动环化”(intron-pair-driven circularization)模型,除此之外还有一种模型是“套索驱动环化”(lariat-driven circularization)模型),这些序列在跨越外显子的内含子区间上可以形成互补序列,所以在转录过程中容易形成茎环结构。环的部分就是外显子,然后在剪接酶的参与下,被剪接成了环形。
另外,cricRNA也存在很多的 可变环形结构 ,这也是由于在基因组中广泛存在的内含子上的Alu序列造成的,不同的Alu序列互补,形成含有不同exon的环形结构。
3. CIRI
这款工具是中科院北京生命科学研究院的赵老师组的工作,今年发表在genome biology上, CIRI: an efficient and unbiased algorithm for de novo circular RNA identification,是一篇方法工具类的文章。
文章中总结了目前从RNA-seq数据中识别circRNA所遇到的问题:
circRNA比其他RNA在细胞中的比例低,一般RNA-seq的实验步骤中,不包括circRNA富集的步骤(例如RNase R treatment,我觉得他们要表到的意思是在RNA-seq中能探测到的circRNA比用RNase R处理过的数据中少),所以在RNA-seq中的circRNA比较低,假阳性也高。
目前的注释文件都是根据线性RNA进行的,所以也不适合circRNA的识别,另外非模式动物的注释信息更少,就更不用谈在那些物种里找circRNA了。
由于RNA测序数据的reads长度差别大,也对检测circRNA工作带来了不便
套索结构和融合基因同circRNA的reads结果类似,不好区分。
文章中总结了从2012年到2014年出现的几种检测circRNA的方法:
Salzman Cell-type specific features of circular RNA expression 用了一个依赖注释信息的方法来检测circRNA,通过搜索已知注释的外显子边界来查找,并在最近的工作中更新了方法,加入了false discovery rate 控制比对的质量分数。这种方法存在上述描述的第二个问题。
Memczak Circular RNAs are a large class of animal RNAs with regulatory potency(也就是第一部分介绍的那个软件)用了GT-AG信号来找寻splicing 位点,也有其他工作用类似的方法筛选micorRNA-sponge 的候选circRNA。这种方法会找不到“长外显子1-短外显子-长外显子2”形成的环形结构,这种结构中一条测序Read上会有三个部分,第一部分序列属于长外显子1,第二部分序列属于短外显子,第三部分序列属于长外显子2。 Memczak的方法只是把一条序列切成两部分,这种算法会把“长外显子1-短外显子-长外显子2”丢掉,或者识别成“长外显子1-长外显子2”。
Jeck Circular RNAs are abundant, conserved, and associated with ALU repeats 采用了比较的方法,比较没有经过RNase处理和经过RNase处理的序列的结果,用来确定潜在的circRNA,消除假阳性。这种方法在富集circRNA阶段会有系统误差。
说了那么多其他人的工作,赵老师组的工作是采用sam格式中的CIGAR值进行分析的,从sam文件中扫描PCC信号(paired chiastic clipping signals)。 CIGAR值在junction read的特征是xS/HyM或者xMyS/H,其中x,y代表碱基数目,M是mapping上的,S是soft clipping,H是hard clipping。
对于单外显子成环,或者“长外显子1-短外显子-长外显子2”形成的环形结构,CIGAR值应该是xS/HyMzS/H以及(x+y)S/HzM或者xM(y+z)S/H,CIRI软件可以很好的将这两种情况分开。
对于paired-end reads,CIRI算法会考虑一对reads,其中一条可以mapping到circRNA上,另一条也需要mapping到circRNA的区间内。
对于splicing 信号(GT,AG) CIRI也会考虑其他弱splicing 信息例如(AT-AC),算法会从GTF/GFF文件中抽取外显子边界位置,并用已知的边界来过滤假阳性。
CIRI方法消耗的内存比较大,我跑了个12G的sam文件,内存用了20G。
方法比较
从一开始的biostar链接中就可以看到不同方法的差异非常大,我测试了一个实际数据,从第一个软件有126163行结果,第二个软件只有327行,第三个软件有687行。"
(以上“”内为网页引用文字)
再回到软件对比介绍
然后看看对比情况:
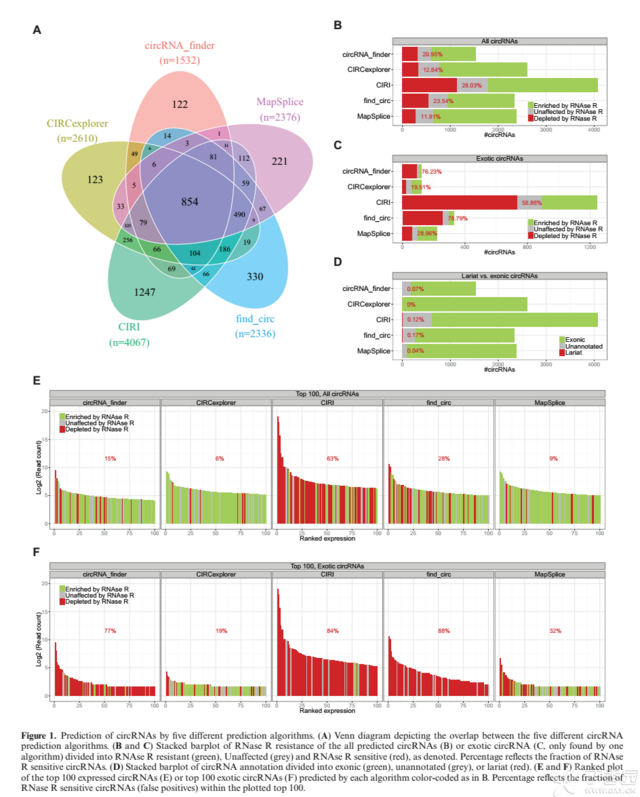
从这个结果可以发现,各个软件都有自己的运用方法,而且都不同,但是可以找到一些交集,可以看上面的维恩图。所以这也是采用多个软件取交集的原因,会提高可信度。
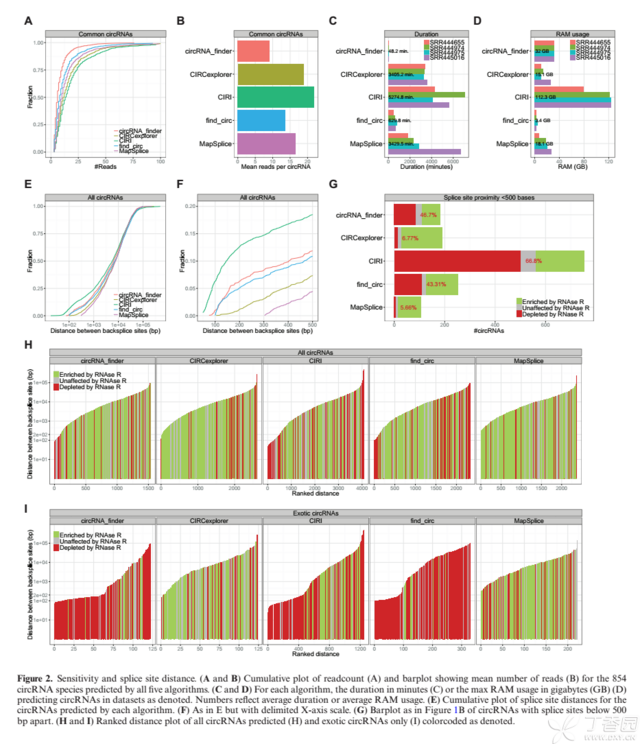
各个软件具体优劣:
最后是联合应用软件预测效果:
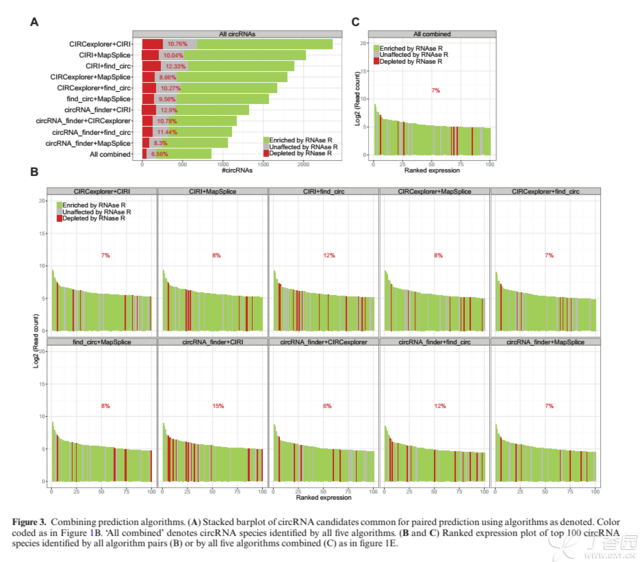
研究者发现:combining any two algorithms would greatly decrease the false positive rate and in general strengthen the output quality.
文章引用之丁香园论坛
文献下载|downloadComparison of circular RNA prediction tools
我来补充一下第二个软件的作者信息,通讯作者为中科院上海生命科学研究院计算生物所的杨力研究员,第一作者为张晓鸥博士,这是他的毕业论文。