







转载自:大土豆力|生信菜鸟团
《2018研究前沿》中,circRNA继去年荣登生物科学TOP10热点榜单后又荣登新兴前沿榜单。自2012年以大数据形式发掘circRNA信息至今,已积累了大量的circRNA生信分析方法及实验验证手段。近期的主题将从数据获取方式、数据挖掘等几个点展开讨论,以期为实验设计提供参考。
前言
从circRNA特殊的拓扑结构来看,head-to-tail junction能够作为检索circRNA的唯一直接证据,但是大约只有0.1%的reads覆盖这个区域,因此在大多数鉴定中低表达的circRNA会被忽略,而这些会限制circRNA的进一步研究。那么,如果你的实验需要尽可能的多获得数据进行分析,应该怎么做呢?我们来看一篇文章。

主要研究方法
数据材料
样本来自于十个病人宫颈癌组织和正常组织及血液。
microarray
首先于circBase下载87935条circRNA序列,将circRNA序列的3‘和5’尾巴组装后获得20nt或者30nt序列,挑取包含反向剪切位点(back splice site)序列作为最终的探针,并以探针 (Control Grid Name: IS-180880-4-V2_4x180K_GX_EQC_201000210)作为参照。
RNA-seq
去核糖体、线性RNA Illumina Hiseq-X10 平台PE测序。
生信分析
实验验证环状RNA
包括RT-PCR,PCR以及sanger测序。
基因共表达分析
4919差异表达的circRNA × 31 680线性RNA探针逐一计算 Pearson correlation coefficient ,≥ 0.9认定为相关基因,DAVID v6.8 用于GO分析。
组织和血液样本的综合分析及Kaplan-Meier分析
GraphPad PRISM 6.0c用于组织和血液样本circRNA相关表达量的归一化,Kaplan-Meier用于估计生存函数。
主要结果
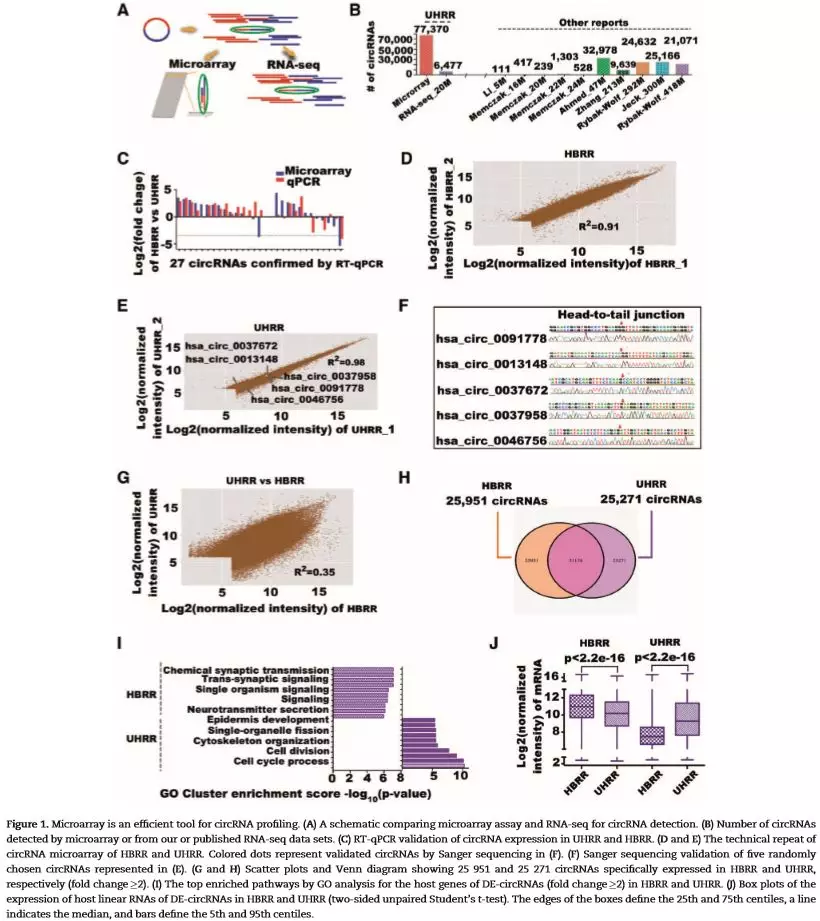
芯片circRNA的识别效率远远高于RNA-seq
在RNA-seq项目中,大于99%的数据都在鉴定中被浪费了,也就是说50 million的测序reads中大约只有20 866–65 594 reads包含接头序列;而microarray由于特殊的探针设计能够几乎100%的是被circRNA (Fig.1)。而在RT-qPCR实验验证过程中,microarry 74%(20 of 27) (Fig.1C)与RNA-seq 75%(21 of 28)基本接近;并随后相关性比较及Sanger测序验证microarray获得circRNA的正确率(Fig.1D,E,F)。
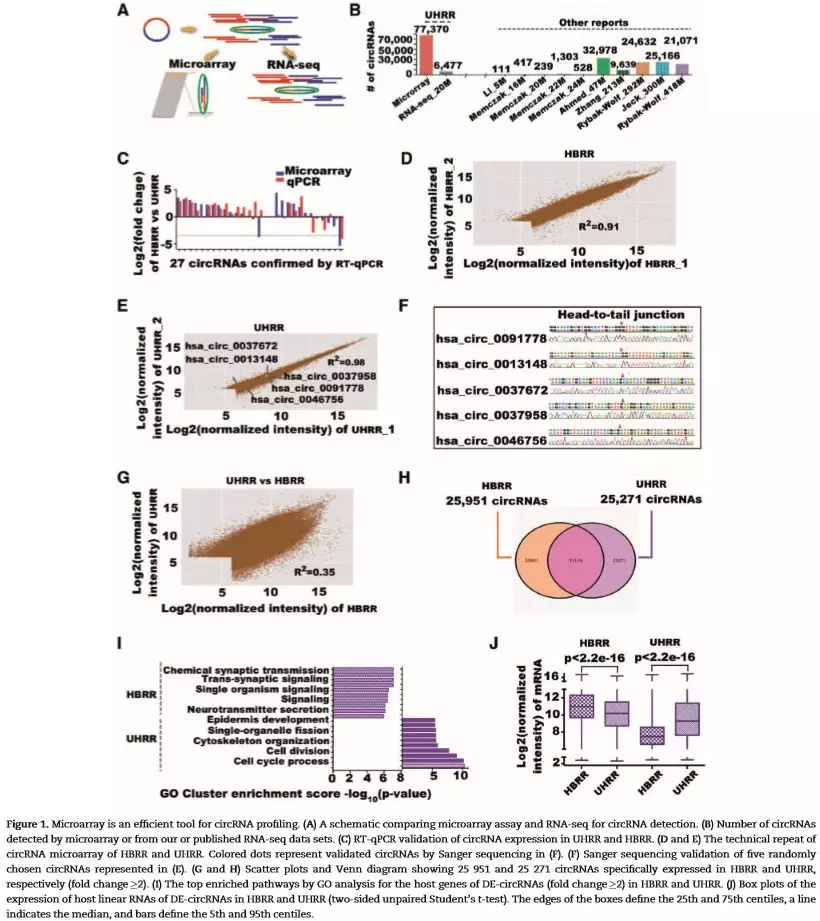
芯片数据能够探测来源基因不表达情况下的circRNA以及弱信号的circRNA
根据来源基因分析,作者发现了1146条microarray获得circRNA的来源基因并没有变动(FPKM <0.01),相同的现象也出现在RNA-seq中,并通过RT-qPCR验证(Fig.2)。同样的方法验证了弱信号的circRNA。
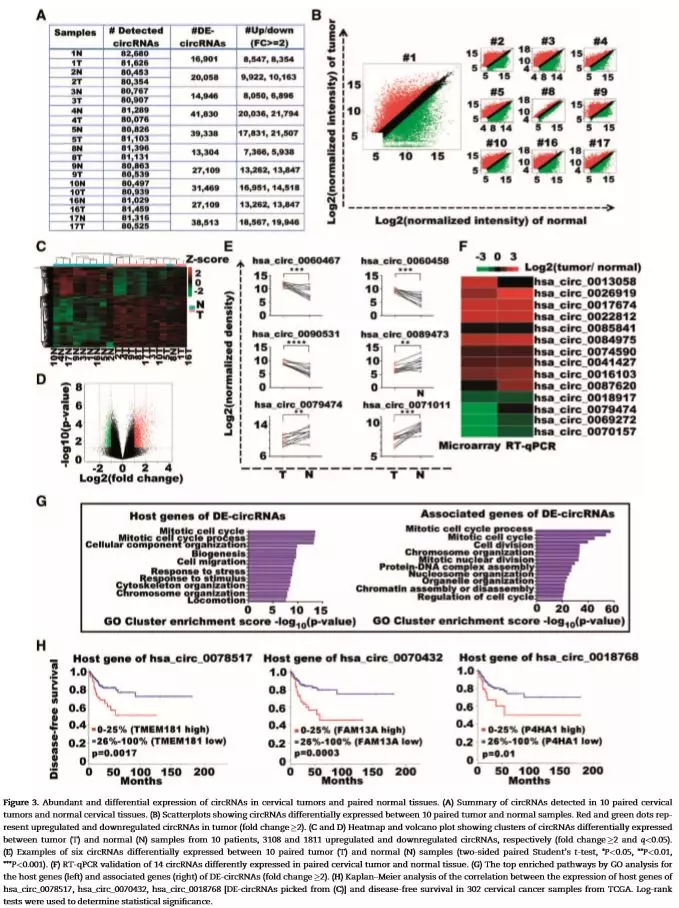
芯片数据对不同组织特异表达的circRNA的识别验证及分析
作者发现10位患者中宫颈肿瘤和正常宫颈组织中检测到平均数量为80 988个circRNA,每位患者样本中平均包含25 749个circRNA差异circRNA (倍数变化≥2) (Fig.3A,B)。对差异显著的circRNA通过GO功能注释发现,其都与癌症高度相关(Fig.3C,D,G)。RT-qPCR验证了78.6%(11 of 14)的芯片差异表达circRNA(Fig.3F)。随后作者通过CLR (circular to linear RNA)的计算,比较了circRNA与线性mRNA之间的表达关系,发现多的circRNA与mRNA的表达并不相关。而circRNA能够来源于同一个基因座,这些circRNA中37.8%表现出不同的趋势。
血液样本中的circRNA芯片的应用及差异分析
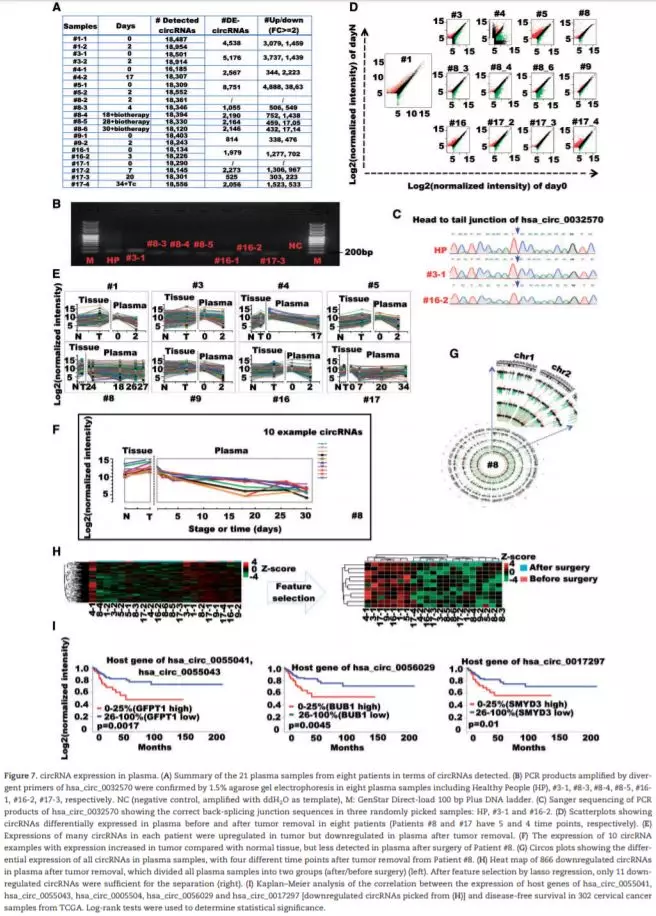
8位癌症患者的21份血浆样品中检测到平均数量为18 293个circRNA(Fig(.7A),并对其中一条序列hsa_circ_0032570进行PCR扩增验证(Fig.7B,C)。最后作者调查了手术前后血液中差异表达的circRNA以探究它们是否参与癌症过程。结果显示,约2787条差异表达的circRNA (fold change ≥2) (Fig.7A,D),通过聚类分析获得了866个下调的circRNA (Fig.7H),进一步Kaplan-Meier分析发现,患者具有差的无病生存期时 (poor disease-free survival),74个circRNA中48个的来源基因具有高表达水平(Fig.7I)。
小结
作者从数据的角度比较两者的差异,仅有0.1%的RNA测序数据中包含circRNA的接头序列(junctions),而根据head-to-tail junction设计的特异探针,可以接近100%获得探测信号用来检测circRNA。文章从多个方面对比了芯片与测序识别circRNA的结果,论证了芯片在circRNA识别上的优势,比如芯片探针受反向剪切序列的噪音干扰影响较弱且具有高特异性;而通过RT-qPCR实验也验证了大部分芯片鉴定的circRNA(78.6%)可信,且芯片识别的circRNA都可以在circBase上有记录。随后作者比较了肿瘤切除前后血液中的circRNA的变化,并获得了一批具有检测生物标记物意义的circRNA。
但是实验对象围绕癌症组织样本及血液,并未对其他的生物组织进行探究。其次,增加测序深度有助于提高RNA测序的识别效率且可以的到更多新circRNA信息,但是由于芯片探针的设计,无法获得新circRNA。因此microarry作为获取的circRNA的方法在特定的场景下具有较大优势。
主要参考文献
[1] Microarray is an efficient tool for circRNA profiling[J]. Briefings in Bioinformatics, 2018, 17(11):241.

来第一个抢占沙发评论吧!